Introduction
EGFR tyrosine kinase inhibitors (TKIs) revolutionized the treatment of lung cancers.1 At that time, we were mainly focused on small genetic modifications. Not- withstanding, in 2007, a chromosomal rearrangement involving ALK was discovered, which was followed by publication of the activity of the ALK inhibitor crizotinib, with high response rates.2,3 The evolution of genomic analysis led to the discovery of novel onco- genic fusion genes such as ROS1 and RET.
In 1985, Takahashi and colleagues4 first described a new transforming gene that appeared to be activated by the recombination of 2 unlinked human DNA segments, possibly by co-integration during transfection of NIH 3T3 cells with human lymphoma DNA, which was designated RET (rearranged during transfection).
The Biology of RET
RET is a proto-oncogene localized in the pericentromeric region of chromosome 10q11.2, which encodes the protein RET, a receptor tyrosine kinase (RTK). RET undergoes alternative splicing of 3’ exons to generate 3 protein isoforms: RET9, RET32, and RET51, which differ at their carboxy terminal amino acids number. RET has 3 domains: a large extracellular domain, a transmembrane region, and an intracellular kinase domain. It is the only RTK with 4 cadherin-like domains in its extracellular region. RET is the signaling receptor for the glial cell-derived neurotrophic factor (GDNF) family of ligands (GFLs): GDNF, neurturin, persephin, and artemin.5 Unlike other RTKs, downstream signaling requires co-receptors that are tethered at the lipid rafts (cholesterol-rich membrane subdomains). Although there can be some crosstalk, each GFL interacts primarily through its specific co-receptor, represented by 4 GDNF family receptor-alpha (GFR-α) 1-4. Upon binding of GFLs to GFR-alpha1-4 complex, RET dimerization and autophosphorylation stimulate multiple downstream pathways, including RAS-MAPK, PI3K-AKT, and STAT3.6,7 These signs play a key role in kidney and nervous system development, neuronal survival and differentiation, and maintenance of spermatogonial stem cells.
RET receptor is expressed in several neural and neuroendocrine cell lineages, such as the thyroid C cells and adrenal chromaffin cells. RET loss-of-function mutations give rise to Hirschsprung disease and congenital abnormalities of the kidney and urinary tract, while RET gain-of-function mutations result in aberrant activation of the receptor; they are pathognomonic in patients with multiple endocrine neoplasia type 2 (MEN2). Both germline and somatic RET mutations represent an important step of medullary thyroid carcinoma onco- genesis. At the same time, somatically occurring RET rearrangements occur in 20% to 40% of papillary thyroid carcinoma.8 The increasing use of new techniques, such as genomic sequencing and transcriptome analysis, has led to the identification of chromosomal rearrangements in other cancers.
Chromosomal rearrangements involving RET are frequently found in irradiation-induced papillary thyroid carcinoma.5
RET and Lung Cancers
In 2012, Ju and colleagues9 first reported on a 33-year- old never-smoker patient with lung adenocarcinoma with a novel fusion gene between KIF5B and the RET proto-oncogene caused by a pericentric inversion of 10p11.22 – q11.21. KIF5B contains a coiled-coil domain functioning as a dimerization unit, which activates the oncogenic tyrosine kinase domain of RET by autophosphorylation after homodimerization. The RET kinase domain portion is preserved in all kinase fusions, despite the breakpoint leaving downstream intracellular kinase activity intact.
The transformation potential of RET fusions has been reported in Ba/F3 cells and LC-2/ad (human adenocarcinoma cell-line), while anchorage-independent cell proliferation has also been shown in NIH3T3 cells.10 The mutually exclusive nature of the RET fusions and other oncogenic alterations suggests that the KIF5B-RET fusion is a driver mutation. However, Kim and colleagues11 reported the co-occurrence of EGFR or KRAS mutations in KIF5B-RET rearranged lung adeno- carcinoma, and RET rearrangement was also reported in patients with EGFR-mutated lung adenocarcinoma who had progressed on TKI therapy.12
A variety of breakage points have been identified within the KIF5B locus, which is the most common fusion partner gene. More importantly, several other RET fusion partner genes have been identified: CCDC6 (coiled-coil domain containing 6), CUX1 (cutlike-homeobox 1), TRIM33 (tripartite-motif containing 33), NCOA4 (nuclear-receptor coactivator 4), KIAA1468, KIAA1217, CLIP1 (CAP-Gly domain containing linker protein 1), ERC1 (ELKS/Rab6- interacting/CAST family member 1), and MYO5C (myosin 5C), among others. Importantly, all of these fusion partners contain coiled- coil domains that are believed to mediate ligand-independent dimerization and constitutive activation of RET.10,13-16
To date, several cancer genome sequencing studies have discovered RET fusions in 1% to 2% of unselected lung cancers, which might be higher in the pan-negative population (negative for all known oncogenic driver mutations).14,17 Several studies have tried to elucidate the clinicopathological characteristics of RET-rearranged lung cancers. Most of the tumors are adenocarcinoma, but some cases involve other histological types, such as adenosquamous carcinoma. The tumors were significantly more common in younger patients and tended to occur in never-smokers and light smokers. The RET-rearranged lung adenocarcinomas are mostly well or moderately differentiated cancers and are thyroid transcription factor 1 (TTF-1) positive; the predominant growth pattern is very heterogeneous.18-21 Interestingly, Lee and colleagues22 reported that the mucinous cribriform pattern was more frequent with CCD6-RET–positive tumors (4/5, 80%), whereas the solid signet-ring cell pattern was present in 3 of 6 (50%) of the KIF5B-RET–positive tumors.
Takeuchi and colleagues13 showed results similar to the aforementioned ones: that the frequency of mucinous cribriform carcinoma was significantly higher in the kinase-fusion–positive group (ALK, ROS1 and RET) of tumors than in the fusion-negative adenocarcinomas. Conversely, the mucinous cribriform pattern was infrequently observed (13.6%) in a Japanese cohort of 22 cases selected from resected specimens at the National Cancer Center, Tokyo.23 Unlike in non–small cell lung cancer (NSCLC), there are some reports of RET gain of-function point mutations in small cell lung cancer (SCLC). Dabir and colleagues24 identified an activating M918T RET somatic mutation in a metastatic small–cell lung cancer (SCLC) tumor specimen, which is among the most highly transforming RET mutations in vitro and leads to a severe clinical MEN2B phenotype.
It is interesting that RET rearrangements develop with a large prevalence in radiation- induced thyroid cancers. Furthermore, exposure to radon is a major risk factor for developing lung cancers. Thus, RET fusions may represent a genetic mechanism of radiation-induced lung adenocarcinoma, but further studies are needed.25
There is no gold-standard technique to detect RET gene fusions, and most studies use multiple techniques, such as whole-genome and transcriptome sequencing, RNA sequencing, reverse transcription polymerase chain reaction (RT-PCR), fluorescence in situ hybrid- ization (FISH), and immunohistochemistry (IHC). Although normal lung tissue shows low RET expression, IHC is not a reliable method to detect overexpressed RET because staining can vary and the immunoreactivity of available antibodies is weak. Overall, a combined strategy of RT- PCR and FISH, with dual color break-apart probe, is an effective tool for detection of RET chromosomal rearrangements. Reverse transcription polymerase chain reaction alone is usually insufficient to detect new partners or isoforms; there- fore, FISH may be better in terms of sensitivity.26 More recently, broad hybrid, capture-based, next-generation sequencing (NGS) was able to identify genomic alterations in 65% of tumors from never- or light-smokers with lung cancers that had previously been deemed free of genomic alterations by the aforementioned types of non-NGS testing. Therefore, NGS should be considered, if feasible.27
Targeting RET
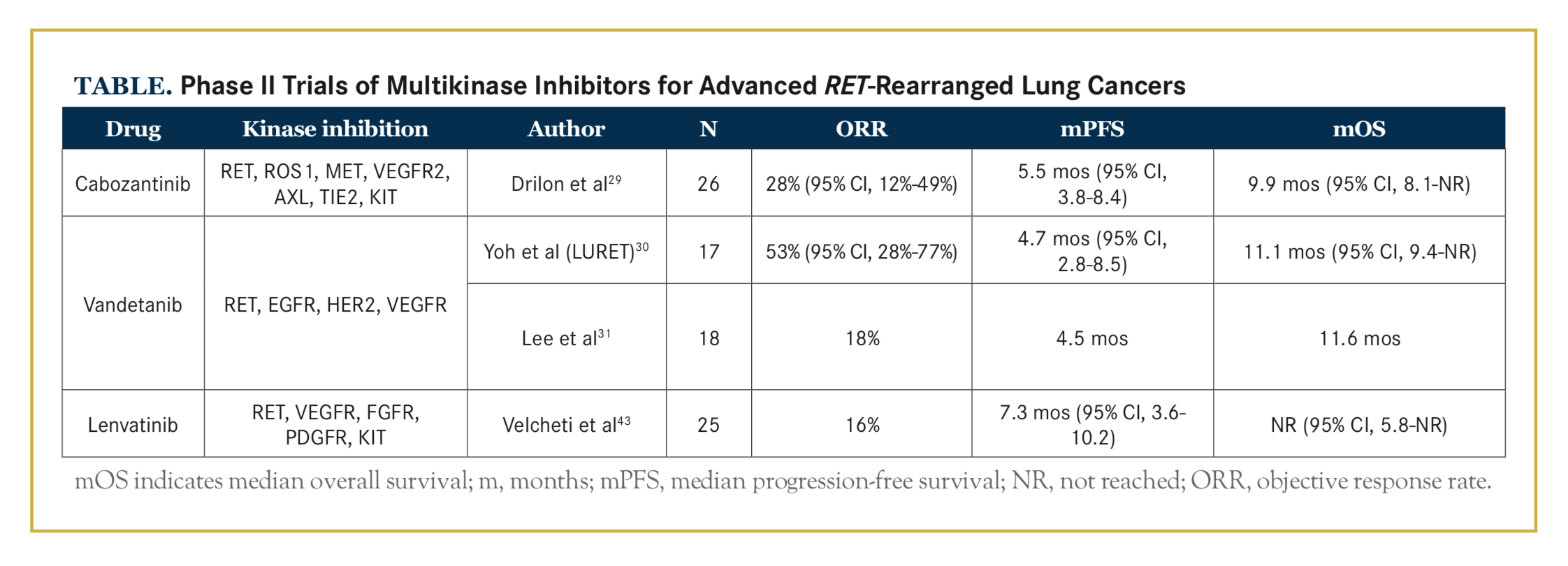
Several commercially available multikinase inhibitors, such as vandetanib (Caprelsa), cabozantinib (Cabometyx), sorafenib (Nexavar), sunitinib (Sutent), lenvatinib (Lenvi- ma), ponatinib (Iclusig), dovitinib (TKI-258), and alectinib (Alecensa), have activity against the RET kinase. In 2013, Drilon et al28 first reported the response to a RET inhibitor, cabozantinib, in patients on a prospective, molecularly enriched trial for RET-positive lung cancers, and in 2016, they published the first stage of a phase II study with 25 cases29 (Table). The most common grade 3 treatment-related adverse events (TRAEs) were lipase elevation, increased levels of alanine aminotransferase and aspartate aminotransferase, decreased platelet count, and hypophosphatemia. Seventy-three percent of patients required cabozantinib dose reduction, most commonly due to pal- mar-plantar erythodysesthesia, fatigue, and diarrhea.
Subsequent reports from 2 phase II trials testing the effect of vandetanib on RET-positive lung cancers showed discordant results, which may be explained by differences in patient selection and choice of assay30,31 (Table). The most common AEs with vandetanib were hyperten- sion, diarrhea, rash acneiform, dry skin, prolonged QT corrected interval, anorexia, and increased creatinine.
Twenty-one percent of patients required vandetanib discontinuation, most commonly due to rash and pneumonitis, and 81% required dose reductions due to rash and hypertension.
Lenvatinib showed clinical benefits in patients with RET-rearranged lung adenocarcinomas, with a disease control rate of 76%, according to a phase II study presented at the 2016 European Society for Medical Oncology Congress. The most commonly reported TRAEs were hypertension, nausea, anorexia, diarrhea, and proteinuria.
All of the aforementioned drugs are multikinase inhibitors with activity against advanced RET-rearranged lung cancers. The objective response rates (ORRs) were modest, but greater than with single-drug chemotherapy or single-drug immunotherapy, after progression on initial platinum doublet treatment in unselected patients with advanced NSCLC. Although clinically meaningful benefit was seen (Table), their activity was lower than that shown with EGFR and ALK inhibitors. These multikinase inhibitors are much more effective at inhibiting VEGFR, EGFR, and KIT than RET, which explains the high rate of off-target dose-limiting toxicities leading to frequent dose reductions and drug discontinuations. Hypertension and proteinuria, both commonly reported, can be related to VEGFR inhibition, while rash acneiform and diarrhea can be due to EGFR inhibition, and skin hypopigmentation and marrow suppression are related to KIT inhibition.
Alectinib, a known inhibitor of ALK, was shown to inhibit RET kinase activity (IC50 = 4.8 nmol/L) and the growth of RET fusion–positive cells by suppressing RET phosphorylation.32 In addition, alectinib showed kinase inhibitory activity against RET gatekeeper mutations (RET V804L and V804M). Lin and colleagues33 described 4 patients with advanced RET-rearranged lung cancers who were treated with alectinib. In total, 2 of 4 patients had overall responses, with durations of therapy of 6 months and more than 5 months. Given its more favorable safety profile, alectinib may be dosed more effectively to target RET, and it can represent an alternative to multikinase inhibitors.
More-specific RET inhibitors, with improved potency and reduced toxicity, are currently being investigated in the clinical and preclinical settings. Early-phase clinical trials of RXDX- 105, a RET and BRAF inhibitor, which spares VEGFR2/KDR and VEGFR1/FLT, have been launched. A patient with advanced RET-rearranged lung cancer had a rapid and sustained response to RXDX-105 in both intracranial and extracranial dis- ease.34 Other RET-specific inhibitors in development include LOXO-292 and BLU-667, which are both potent VEGFR-sparing RET inhibitors with specificity for RET and predicted resistant mutants. Of note, different sensitivities to RET inhibitors among different RET fusion forms are still unknown and need further study.
As in the case of other oncogene-driven lung cancers, resistance to RET inhibition is likely to emerge. We speculate that resistance to RET inhibition from the available multikinase inhibitors may be mediated more frequently by bypass signaling mechanisms than by RET-resistant mutations, because lower activity against RET exerts less selective pressure over the RET pathway. Also, RET-rearranged lung cancers might rely on alternative signaling pathways, and combination treatment may represent an alternative in the future.35,36
As with ALK- and ROS1-rearranged lung cancers, durable benefits with pemetrexed-based therapies in RET-rearranged lung cancers were seen. Drilon and colleagues37 retrospectively evaluated 104 patients with RET-rearranged lung cancers who received treatment with pemetrexed alone or in combination. Patients had a median PFS of 19 months (95% CI, 12-not reached) and an ORR of 45%. One might expect lower response rates to immunotherapy in RET-positive lung cancers, in accordance with other oncogene-driven lung cancers. A recent meta-analysis to assess the role of immune checkpoint inhibitors as second-line therapy in EGFR-mutant, advanced NSCLC showed that immunotherapy does not improve OS over docetaxel in this population. Gainor and colleagues38 observed a low ORR in a cohort of 58 patients with EGFR-mutant and ALK-positive lung cancer treated with a PD-1/PD-L1 inhibitor. Also, poor results with checkpoint blockade in patients with MET exon 14‒mutant lung cancer were presented at the 2017 American Society of Clinical Oncology Annual Meeting. While PD-L1 expression was found in RET-rearranged lung cancers, the potential efficacy of checkpoint blockade in this population has not been tested so far.
Conclusions
RET-rearranged lung cancers represent a small subset of lung adenocarcinomas with clinicopathological features similar to those of other rearrangement-driven lung cancers. Given the low frequency of these cancers, collaboration among various international research centers can generate meaningful knowledge about them. A global, multicenter network of thoracic oncologists (RET registry) identified 165 patients with RET-rear- ranged lung cancers and has recently published the resultant data.39
Several multikinase inhibitors have shown activity and clinical benefit with RET-rearranged lung adeno- carcinomas, which raises the question of whether this activity might be related to VEGF inhibition solely, as these drugs have shown increased response rates with unselected lung cancers after platinum-based chemo- therapy.40,41 Dose reductions, likely related to off-target toxicities due to concomitant inhibition of non-RET kinases, prevent the delivery of optimal dosage. In addition, RET-rearranged lung cancer may also harbor concomitant genetic alterations that can decrease the likelihood of response to available RET inhibitors.
We eagerly await the new specific RET inhibitors, which, encouragingly, have less off-target toxicity and more potency. Recent advances in diagnostics should facilitate the identification of patients who will potentially benefit. Unbiased approaches using next-generation sequencing, including whole-genome sequencing, sequencing after capture of selected regions of RNA or DNA encompassing the relevant breakpoints in RET, or transcriptome sequencing of RNA, may be the best methodologies for the detection of RET chromosomal rearrangements in lung adenocarcinoma.42 This approach supports the conduct of “basket trials,” early-phase studies of novel targeted therapies specifically in patients whose tumors harbor the putative oncogenic target.
Author affiliations: Drs Santini and Katz are from the Hospital Sírio Libanes, Sao Paulo, Brazil, and Dr Santini is also from the Instituto do Cancer do Estado de Sao Paulo.
Address correspondence to: Fernando C. Santini, MD, Hospital Sírio Libanes, Oncology Center, Rua Adma Jafet 91, Sao Paulo, SP, 01308-050, Brazil. Tel: +55 (11)33940200; E-mail: [email protected].
Financial disclosures: The authors report that they have no relevant financial relationships to disclose.
References
- Tsao M-S, Sakurada A, Cutz J-C, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133-144. doi: 10.1056/NEJMoa050736.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma ki- nase inhibition in non- small-cell lung cancer [published correction appears in ]. N Engl J Med. 2010;363(18):1693-1703. doi: 10.1056/ NEJMoa1006448.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561-566. doi: 10.1038/nature05945.
- Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581-588.
- Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer.2014;14(3):173-186. doi: 10.1038/nrc3680.
- Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16(4- 5):441-467. doi: 10.1016/j.cytogfr.2005.05.010.
- Qian Y, Chai S, Liang Z, et al. KIF5B-RET fusion kinase pro- motes cell growth by multilevel activation of STAT3 in lung cancer. Mol Cancer. 2014;13:176. doi: 10.1186/1476-4598-13-176.
- Eng C. RET proto-oncogene in the development of human cancer. J ClinOncol.1999;17(1):380-393. doi: 10.1200/JCO.1999.17.1.380.
- Ju YS, Lee WC, Shin JY, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22(3):436-445. doi: 10.1101/gr.133645.111.
- Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375-377. doi: 10.1038/nm.2644.
- Kim JO, Lee J, Shin JY, et al. KIF5B-RET Fusion gene may coincide oncogenic mutations of EGFR or KRAS gene in lung adenocarcinomas. Diagn Pathol. 2015;10:143. doi: 10.1186/s13000-015-0368-z.
- Klempner SJ, Bazhenova LA, Braiteh FS, et al. Emergence of RET rearrangement co- existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer. 2015;89(3):357-359. doi: 10.1016/j.lungcan.2015.06.021.
- Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378-381. doi: 10.1038/nm.2658.
- Li F, Feng Y, Fang R, et al. Identification of RET gene fusion by exon array analyses in “pan-negative” lung cancer from never smokers. Cell Res. 2012;22(5):928-931. doi: 10.1038/cr.2012.27.
- Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382-384. doi: 10.1038/nm.2673.
- Lee MS, Kim RN, I H, et al. Identification of a novel partner gene, KIAA1217, fused to RET: functional characterization and inhibitor sensitivity of two isoforms in lung adenocarcinoma. Onco- target. 2016;7(24):36101-36114. doi: 10.18632/oncotarget.9137.
- Kohno T, Tsuta K, Tsuchihara K, et al. RET fusion gene: translation to personalized lung cancer therapy. Cancer Sci. 2013;104(11):1396-1400. doi: 10.1111/cas.12275.
- Wang R, Hu H, Pan Y, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30(35):4352- 4359. doi: 10.1200/JCO.2012.44.1477.
- Lin C, Wang S, Xie W, et al. The RET fusion gene and its correlation with demographic and clinicopathological features of non-small cell lung cancer: a meta-analysis. Cancer Biol Ther. 2015;16(7):1019-1028. doi: 10.1080/15384047.2015.1046649.
- Pan Y, Zhang Y, Li Y, et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer. 2014;84(2):121-126. doi: 10.1016/j.lungcan.2014.02.007.
- Michels S, Scheel AH, Scheffler M, et al. Clinicopathological characteristics of RET rearranged lung cancer in European patients. J Thorac Oncol. 2016;11(1):122-127. doi: 10.1016/j.jtho.2015.09.016.
- Lee SE, Lee B, Hong M, et al. Comprehensive analysis of RET and ROS1 rearrangement in lung adenocarcinoma. Mod Pathol. 2015;28(4):468-479. doi: 10.1038/modpathol.2014.107.
- Tsuta K, Kohno T, Yoshida A, et al. RET-rearranged non-small- cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer. 2014;110(6):1571-1578. doi: 10.1038/bjc.2014.36.
- Dabir S, Babakoohi S, Kluge A, et al. RET mutation and expres- sion in small-cell lung cancer. J Thorac Oncol. 2014;9(9):1316-1323. doi: 10.1097/JTO.0000000000000234.
- Dacic S, Luvison A, Evdokimova V, et al. RET rearrange- ments in lung adenocarcinoma and radiation. J Thorac Oncol. 2014;9(1):118-120. doi: 10.1097/JTO.0000000000000015.
- Go H, Jung YJ, Kang HW, et al. Diagnostic method for the detection of KIF5B-RET transformation in lung adenocarcinoma. Lung Cancer. 2013;82(1):44-50. doi: 10.1016/j.lungcan.2013.07.009.
- Drilon A, Wang L, Arcila ME, et al. Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin Cancer Res. 2015;21(16):3631-3639. doi: 10.1158/1078-0432.CCR-14-2683.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3(6):630-635. doi: 10.1158/2159-8290.CD-13-0035.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET- rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016;17(12):1653-1660. doi: 10.1016/S1470- 2045(16)30562-9.
- Yoh K, Seto T, Satouchi M, et al. Vandetanib in patients with previously treated RET- rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med. 2017;5(1):42-50. doi: 10.1016/S2213- 2600(16)30322-8.
- Lee SH, Lee JK, Ahn MJ, et al. Vandetanib in pretreated patients with advanced non- small cell lung cancer-harboring RET rearrangement: a phase II clinical trial. Ann Oncol. 2017;28(2):292- 297. doi: 10.1093/annonc/mdw559.
- Kodama T, Tsukaguchi T, Satoh Y, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther. 2014;13(12):2910- 2918. doi: 10.1158/1535-7163.MCT-14-0274.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET- rearranged non-small cell lung cancer. J Thorac Oncol. 2016;11(11):2027-2032. doi: 10.1016/j.jtho.2016.08.126.
- Li GG, Somwar R, Joseph J, et al. Antitumor activity of RXDX- 105 in multiple cancer types with RET rearrangements or muta- tions. Clin Cancer Res. 2017;23(12):2981-2990. doi: 10.1158/1078-0432.CCR-16-1887.
- Nelson-Taylor SK, Le AT, Yoo M, et al. Resistance to RET-in- hibition in RET-rearranged NSCLC is mediated by reactivation of RAS/MAPK signaling. Mol Cancer Ther. 2017;16(8):1623-1633. doi: 10.1158/1535-7163.MCT-17-0008.
- Chang H, Sung JH, Moon SU, et al. EGF induced RET inhib- itor resistance in CCDC6- RET lung cancer cells. Yonsei Med J. 2017;58(1):9-18. doi: 10.3349/ymj.2017.58.1.9.
- Drilon A, Bergagnini I, Delasos L, et al. Clinical outcomes with pemetrexed-based systemic therapies in RET-rearranged lung cancers. Ann Oncol. 2016;27(7):1286-1291. doi: 10.1093/annonc/mdw163.
- Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22(18):4585-4593. doi: 10.1158/1078-0432.CCR-15-3101.
- Gautschi O, Milia J, Filleron T, et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multi- center RET Registry. J Clin Oncol. 2017;35(13):1403-1410. doi: 10.1200/JCO.2016.70.9352.
- de Boer RH, Arrieta Ó, Yang CH, et al. Vandetanib plus peme- trexed for the second-line treatment of advanced non-small-cell lung cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2011;29(8):1067-1074. doi: 10.1200/JCO.2010.29.5717.
- Herbst RS, Sun Y, Eberhardt WE, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11(7):619- 626. doi: 10.1016/S1470-2045(10)70132-7.
- Watanabe H, Brooks AN, Meyerson M. Breaking down RET breakpoints in lung adenocarcinoma. J Thorac Oncol. 2014;9(5):590- 592. doi: 10.1097/JTO.0000000000000168.
- Velcheti V, Hida T, Reckamp KL, et al. Phase 2 study of lenvati- nib (LN) in patients (Pts) with RET fusion-positive adenocarcinoma of the lung. Ann Oncol. 2016;27(suppl 6):1204PD. doi: 10.1093/annonc/mdw383.05.





