Introduction
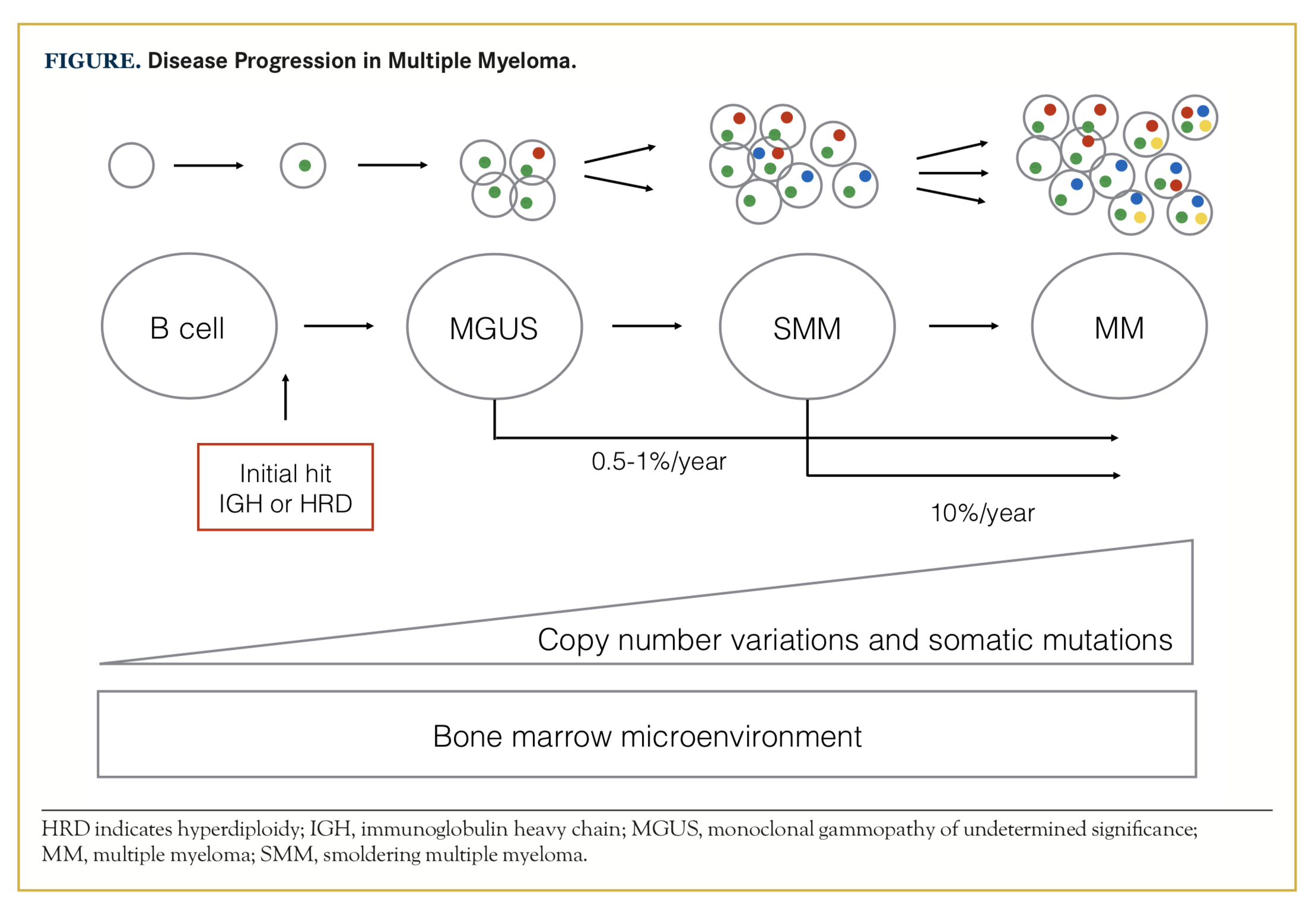
Multiple myeloma (MM) is characterized by proliferation of bone marrow plasma cells and secretion of monoclonal paraprotein or light chains in blood and/ or urine. It is an incurable disease, but novel medications introduced during recent years have led to a significant increase in progression-free survival and overall survival (OS).1,2 MM is consistently preceded by the precursor state of monoclonal gammopathy of undetermined significance (MGUS), which can progress to smoldering MM (SMM) and to MM requiring treatment.3 MGUS exists in 2.4% of the population older than 50 years, and it is more common in African Americans compared with Caucasian whites and Mexican Americans.4 The onset of MGUS starts between age 30 to 40 years, and starts an average 10 years earlier in blacks. Approximately 0.5% to 1% of patients with MGUS and 10% of patients with SMM progress to MM each year.5,6 The risk of progression is higher in individuals who have a higher M-protein, non-IgG MGUS and a skewed free light chain ratio.5,7 Current risk scoring systems rely mainly on the overall burden of disease, and there is a lack of well-defined biological features that hold prognostic information.
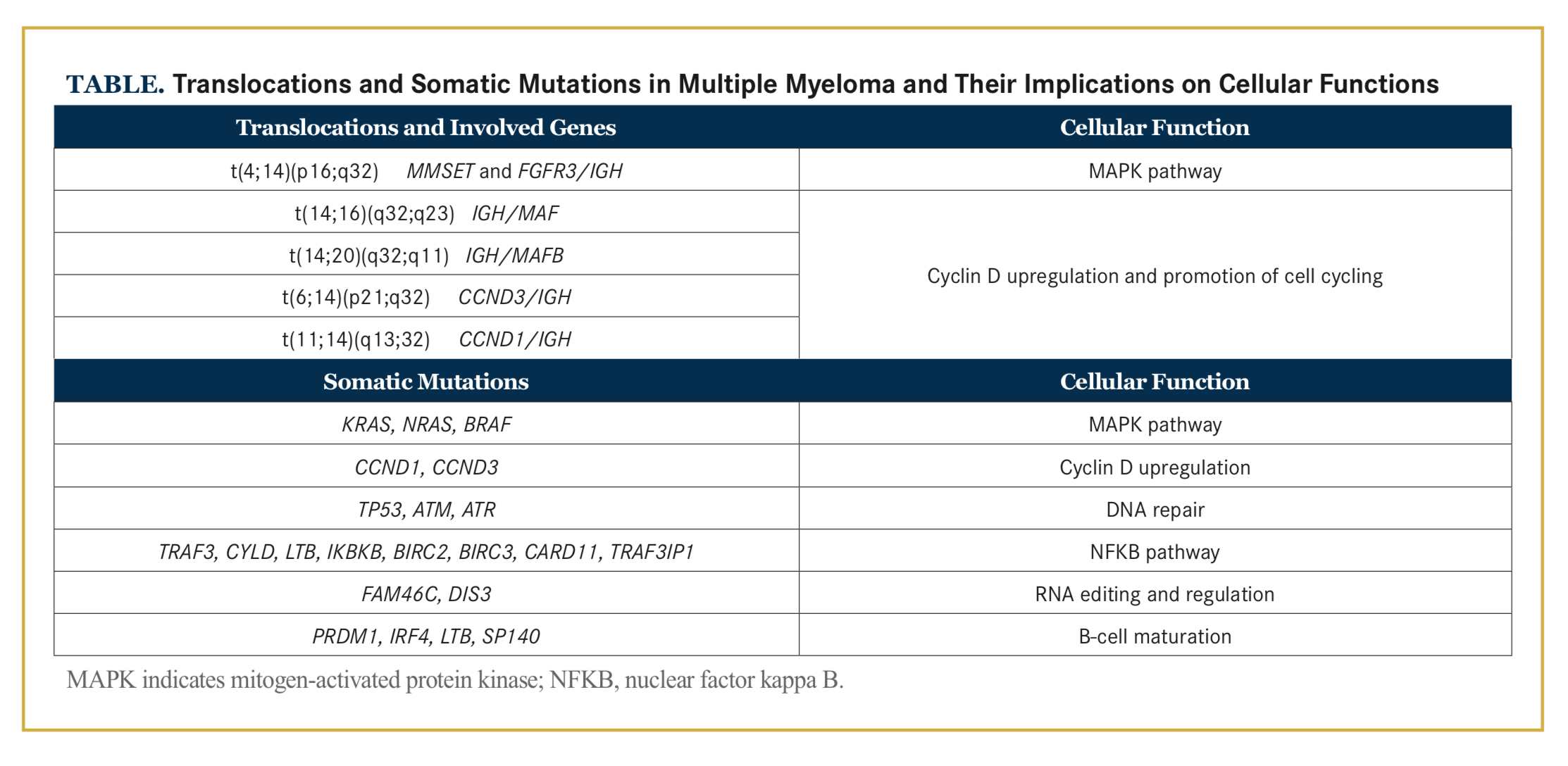
The pathogenesis of MM includes multiple genetic aberrations as well as changes in the bone marrow microenvironment that allow evolution and proliferation of malignant plasma cells. The genetic landscape in MM is complex and includes translocations, copy number variations (CNVs), and somatic mutations that affect several molecular pathways and cellular functions (Table). Additionally, the disease consists of a number of subclones that may vary in size and number through- out the disease course. In this review, we focus on the current knowledge of genomic complexity and disease evolution in MM.
Plasma Cell Development
Plasma cells originate from B lymphocytes produced in the bone marrow. After initial rearrangement of the immunoglobulin heavy chain and light chain genes, the naïve B cells leave the bone marrow expressing immature immunoglobulins on their surfaces. After encountering an antigen, the B cells migrate to the germinal center of the lymph node for further maturation. The maturation process includes somatic hypermutation, which induces point mutations in the variable region of immunoglobulin heavy chain (IGH), resulting in highly specific immunoglobulins and class switch recombination, which enhances the affinity and specificity of the immunoglobulins.8-11 These processes require genetic editing through DNA double-strand breaks in IGH mediated by activation-induced cytidine deaminase (AID), an enzyme belonging to the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) family.8 The maturation process results in production of plasmablasts, which are initial antibody-producing effector cells, and memory B cells. Terminal differentiation of plasmablasts results in long-lived plasma cells with highly specific antibodies.8-10
Neoplastic development of plasma cells in MM occurs through a multistage process involving acquisition of genetic events and deregulation of plasma cells. The microenvironment plays an important role in promoting the expansion of the premalignant and, later, malignant clones.12 As the disease evolves, the neoplastic plasma cells and the subsequent overproduction of immunoglobulin heavy and light chains eventually lead to end-organ damage defined by the CRAB criteria (hyperCalcemia, Renal failure, Anemia, Bone lesions).13
Genetic Landscape
The genetic landscape in MM is complex and involves multiple types of genetic aberrations, such as translocations, CNVs, and somatic mutations.8 Initially, chromosomal aberrations were assessed using metaphase cytogenetics, a technique that requires cell division in vitro, which may be challenging since plasma cells are slow-dividing cells. In addition, some of the more common translocations are cryptic and not captured by conventional cytogenetics. In clinical practice, fluorescence in situ hybridization (FISH) is widely used to capture translocations and CNVs. This technique is labor-intensive, and findings are limited to the specific targeted probes used in each patient.14 With the development of massive parallel sequencing, novel insights into the genomic landscape of MM have been gained.
Chromosomal Translocations and Copy Number Variations
The cytogenetic changes in MM can be broadly divided into 2 groups: IGH translocations and hyperdiploidy. IGH translocations occur during the genetic editing in the germinal center of the lymph node, where occasion- ally the double-strand DNA is aberrantly rejoined.9 The IGH breakpoints tend to fall within certain preferred loci, and these translocations result in the juxtaposition of IGH and an oncogene.15 The oncogene is placed under the strong IGH enhancer and is overexpressed.8,15 The most common IGH translocations are t(4;14), t(6;14), t(11;14), t(14;16), and t(14;20).15,16
Translocation 4;14 results in MMSET and FGFR3 being overexpressed in 100% and 70% to 75% of cases, respectively. Upregulation of MMSET alters epigenetic gene regulation, and FGFR3 encodes for a tyrosine kinase receptor with oncogenic potential when up- regulated or activated.15,17-19 Translocations between chromosome 14 and chromosomes 11 and 6, affecting the partner genes CCND1 and CCND3, respectively, result in upregulation of cyclin D proteins and promotion of cell cycling.19,20 Furthermore, t(14;16) and t(14;20) affect MAF and MAFB, respectively, in which the downstream effects include upregulated expression of a number of genes, including CCND2.19,21-23
Translocations t(4;14), t(14;16), and t(14;20) are considered high-risk aberrations and associated with adverse prognosis.21,24,25 Translocation 11;14 was previously considered to have an overall neutral effect; however, emerging data on t(11;14) implies that it may confer a worse-than-standard-risk prognosis.26
Yet it is important to emphasize that these and other genetic subtypes have been designated as high risk in terms of survival among patients with these genetic markers (vs those without). Because clinical outcomes are highly dependent on the given treatment, in the future, many patients with genetic features that previously were considered to confer high risk will likely have the same outcome as standard-risk patients. Thus, with better therapies the proportion of patients with poor outcomes will become smaller.27
Approximately 45% of patients with MM harbor IGH translocations, while approximately 40% have trisomies of the odd-numbered chromosomes 3, 5, 7, 9, 11, 15, 19, and 21. The mechanism behind hyperdiploidy is less clear, but the acquisition of extra chromosomes is hypothesized to happen during one catastrophic mitosis rather than a step-wise gain of chromosomes.19 IGH translocations and hyperdiploidy are early hits and are both found already at the MGUS stage (Figure), and they are therefore not considered by themselves to be sufficient for development of MM.28,29
Secondary chromosomal events include 17p deletion, gain of 1q, deletion of 1p, and deletion 13q, of which the majority are associated with adverse OS.30,31 Many of the secondary CNVs are subclonal, indicating that they are acquired during the disease course rather than being founder events, in contrast to IGH translocations and hyperdiploidy.32
Approximately 3% to 4% of patients with MGUS and SMM harbor MYC translocations, whereas MYC translocations are found in up to 15% to 20% of patients with newly diagnosed MM.32,33 When including patients who have gains of the MYC locus, up to 30% of patients have events involving MYC, making this one of the most common aberrations in MM.32
Somatic Mutations
The introduction of massive parallel sequencing has allowed high-resolution sequencing of large cohorts yielding important new insights into the genetic landscape of MM. During the past 5 years, mainly through whole-exome sequencing, a number of frequently mutated genes have been reported. The most common mutations are found within the mitogen-activated protein kinase (MAPK) pathway: KRAS, NRAS, and BRAF are mutated in approximately 50% of patients with MM.34-37 Mutations in KRAS and NRAS are mutually exclusive in the majority of cases, but they do coexist in 2% of patients.36 Additional genes that are frequently mutated in MM are FAM46C, TP53, DIS3, IRF4, TRAF3, CYLD, RB1, SP140, LTB, MAX, EGR1, FGFR3 ATM, ATR, and more.35,36,38 Of these, KRAS, NRAS, TP53, BRAF, FAM46C, and DIS3are generally considered to be driver mutations.10 These mutations affect various cell functions, such as MAPK and nuclear factor kappa B signaling, DNA repair mechanisms, and RNA editing.19,35,36,38
The genetic complexity and mutational burden increase as the disease progresses, but there is no genetic profile specific to MGUS, SMM, or MM.10,28,30,39 Disease evolution may occur either through gains of additional mutations or expansion of clones that are already present at an early disease stage, but that initially fall below the level of detection.10,40 Interestingly, Mailankody et al41 found that the overall number of somatic mutations was similar between SMM and MM, but the pattern of mutations was different between the disease stages. In patients with MM, there were more mutations in genes that have been reported as frequently mutated and in driver genes, compared with patients with SMM. Further- more, patients who had a good response to treatment had fewer mutations in these frequently mutated genes compared with those with a poorer response when treated with the modern combination treatment of carfilzomib/lenalidomide/dexamethasone.41
Mutational Processes and Altered Pathway Activity
The overall mutation rate in MM is higher than in other hematologic malignancies, but lower than in many solid tumors.19,42 Several mutational processes and signatures are present in MM, with the most prominent mutational signatures being 1, 2, 5, and 13, which are commonly signatures of aging and AID/APOBEC activity.43 Kataegis, a process of regional clustering of mutations close to trans- location breakpoints, is also present in MM, both in IGH and MYC translocations.38 In 11% to 25% of patients, the partner gene on der(14) in an IGH translocation—CCND1, FGFR3, MAF, or MAFB—is hypermutated.32,38
Thus, cellular pathways and functions can be altered through different mechanisms, which may have additive effects. As an example, TP53 located on 17p may be inactivated through chromosomal arm deletion or inactivated though TP53 mutations.44 Another example may be the MAPK pathway, which is activated through mutations in KRAS/NRAS/BRAF, but can also be activated through translocation involving t(4;14) or FGFR3, leading to increased activity of the tyrosine kinase and downstream upregulation of the MAPK pathway.45 There is also evidence of co-occurrence of gene–gene and gene–CNV aberrations where biallelic events (eg, 17p deletion and TP53 mutation as well as 1p deletion and FAM46C mutation) are associated with a worse prognosis than if only 1 allele is affected.46
Clonal Evolution
In MM, there are, on average, 5 heterogeneous sub- clones, and the disease is thought to progress through Darwinian evolution driven by competing subclones.35,38 As with CNVs, mutations can be clonal or subclonal, and the subclones vary in size and distribution over the disease course in response to clonal competition and treatment.35,38,47,48 Bolli et al39 analyzed longitudinal samples in a subset of patients and described different patterns of progression: linear progression with the same clone present at relapse, branching progression with a new subclone appearing at relapse, or branching progression with a different dominant clone at relapse, with or without the initial dominant clone still present. Moreover, certain high-risk events (eg, 17p deletion, and mutations conferring treatment resistance, such as CRBN) are more common in relapse samples.19,48
Clinical Implications
The clinical staging system is currently based on labora- tory findings (beta-2 microglobulin, lactate dehydroge- nase, and albumin levels) and presence of high-risk IGH translocations and CNVs, t(4;14), t(14;16), or deletion 17p.49 Presence of these aberrations can impact the clinical decision making in favor of more aggressive treatment.24 As more and more targeted drugs are being developed, the mutational landscape will be increasingly important to assess in patients with MM. For instance, the BCL2 inhibitor venetoclax has effect in relapsed/refractory MM harboring t(11;14), and ongoing studies with BRAF and MEK inhibitors are targeting the MAPK pathway for patients with mutations in BRAF, KRAS, and NRAS.50-52 Furthermore, it seems logical to propose that future prog- nostic markers/models—across hematologic malignancies—likely will focus more on the genetic landscape of residual tumor cells posttherapy. Indeed, such observations have already been made in patients with acute myeloid leukemia (AML). Clearance of adverse genetic aberrations 30 days posttherapy has been proposed to be one of the strongest favorable prognostic factors in AML.53 We and others are currently conducting such studies in plasma cell disorders to better define these dynamics in MM.
Summary and Future Perspectives
Detailed interrogation of the genetic landscape of MM and its precursor disease using modern sequencing techniques has revealed a complex genetic landscape, as well as interpatient and intrapatient spatial and temporal heterogeneity. Multiple genetic editing and mutational processes are in place, as well as Darwinian evolution through competing subclones. In this review, we focused on the genomic events of the tumor cells. Nonetheless, the bone marrow microenvironment and the host immune system likely play a large role in the pathogenesis of MM. The understanding of interactions among the microenvironment and the tumor cells is, however, less developed and goes beyond the scope of this review.
Despite the recent advances in genomic events in MM, areas of interest still exist where information is currently limited. First, the majority of sequencing studies have been cross-sectional and included heterogeneous patient populations. Thus, longitudinal studies with serial samples are needed to increase our knowledge on temporal relationships and clonal evolution, both in early disease as well as at relapse. Second, through assessment of gene– gene, gene–CNV, and gene–treatment interactions in the era of modern combination treatments, we will be able to identify distinct molecular profiles and optimize treatment prediction models. As the availability and accuracy of the sequencing techniques and bioinformatic analyses increase, sequencing will be used to identify translocations, CNVs, and mutations and to monitor minimal residual disease, and it will eventually replace conventional cytogenetics and FISH.34,54 With additional high-resolution methods such as circulating tumor cells and cell-free DNA, the possibilities of assessing genomic profiles of plasma cell disorders throughout the disease trajectory will be increasingly accessible.55,56 In summary, accurate and available technologies will further increase our knowledge of molecular driver events. This, in turn, is essential for development of early and targeted treatments to improve patient outcomes in MM.
Author affiliations: Drs Hultcrantz and Landgren are with Memorial Sloan Kettering Cancer Center, Department of Medicine, Myeloma Service, New York, NY, and Dr. Hultcrantz is also with Karolinska University Hospital and Karolinska Institutet, Department of Medicine, Solna, Division of Hematology, Stockholm, Sweden.
Address correspondence to: Malin Hultcrantz, MD, PhD, Myeloma Service, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; Tel: (212) 639-5126; Fax: (646) 227-7116; E-mail: [email protected]
Financial disclosures: The Swedish Research Council (2015-00564) (Hultcrantz), Multiple Myeloma Research Foundation (2015 Research Fellow Award) (Hult- crantz), The Swedish Cancer Society (CAN 2014/1329) (Hultcrantz), and MSKCC Core grant (P30 CA008748) (Hultcrantz, Landgren).
References
- Landgren O, Iskander K. Modern multiple myeloma therapy: deep, sustained treatment response and good clinical outcomes. J Intern Med. 2017;281(4):365-382. doi: 10.1111/joim.12590.
- Kristinsson SY, Anderson WF, Landgren O. Improved long-term survival in multiple myeloma up to the age of 80 years. Leukemia. 2014;28(6):1346-1348. doi: 10.1038/leu.2014.23.
- Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412- 5417. doi: 10.1182/blood-2008-12-194241.
- Landgren O, Graubard BI, Katzmann JA, et al. Racial disparities in the prevalence of monoclonal gammopathies: a population-based study of 12,482 persons from the National Health and Nutritional Examination Survey. Leukemia. 2014;28(7):1537-1542. doi: 10.1038/leu.2014.34.
- Turesson I, Kovalchik SA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance and risk of lymphoid and myeloid malignancies: 728 cases followed up to 30 years in Sweden. Blood. 2014;123(3):338-345. doi: 10.1182/blood-2013-05-505487.
- Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111(2):785-789.
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106(3):812-817.
- Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335-348. doi: 10.1038/nrc3257.
- Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The< generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15(3):160-171. doi: 10.1038/nri3795.
- Dutta AK, Hewett DR, Fink JL, Grady JP, Zannettino ACW. Cutting edge genomics reveal new insights into tumour development, disease progression and therapeutic impacts in multiple myeloma. Br J Haematol. 2017;178(2):196-208. doi: 10.1111/bjh.14649.
- Klein U, Goossens T, Fischer M, et al. Somatic hypermutation in normal and transformed human B cells. Immunol Rev. 1998;162:261-280.
- Balakumaran A, Robey PG, Fedarko N, Landgren O. Bone marrow microenvironment in myelomagenesis: its potential role in early diagnosis. Expert Rev Mol Diagn. 2010;10(4):465-480. doi: 10.1586/erm.10.31.
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi: 10.1016/S1470-2045(14)70442-5.
- Joseph NS, Gentili S, Kaufman JL, Lonial S, Nooka AK. High- risk multiple myeloma: definition and management. Clin Lymphoma Myeloma Leuk. 2017;17S:S80-S87. doi: 10.1016/j.clml.2017.02.018.
- Walker BA, Wardell CP, Johnson DC, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121(17):3413-3419. doi: 10.1182/blood-2012-12-471888.
- Gonzalez D, van der Burg M, Garcia-Sanz R, et al. Immuno- globulin gene rearrangements and the pathogenesis of multiple myeloma. Blood. 2007;110(9):3112-3121.
- Keats JJ, Maxwell CA, Taylor BJ, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. 2005;105(10):4060-4069.
- Santra M, Zhan F, Tian E, Barlogie B, Shaughnessy J Jr. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood. 2003;101(6):2374-2376.
- Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100-113. doi: 10.1038/nrclinonc.2016.122.
- Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaugh- nessy J Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106(1):296-303.
- Stella F, Pedrazzini E, Agazzoni M, Ballester O, Slavutsky I. Cy- togenetic alterations in multiple myeloma: prognostic significance and the choice of frontline therapy. Cancer Invest. 2015;33(10):496- 504. doi: 10.3109/07357907.2015.1080833.
- Hurt EM, Wiestner A, Rosenwald A, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5(2):191-199.
- Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020-2028.
- Sonneveld P, Avet-Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127(24):2955- 2962. doi: 10.1182/blood-2016-01-631200.
- Chng WJ, Dispenzieri A, Chim CS, et al; International Myeloma Working Group. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014;28(2):269-277. doi: 10.1038/leu.2013.247.
- Lakshman A, Moustafa MA, Rajkumar SV, et al. Natural his- tory of t(11;14) multiple myeloma [published online June 27, 2017]. Leukemia. doi: 10.1038/leu.2017.204.
- Landgren O, Rajkumar SV. New developments in diagnosis, prognosis, and assessment of response in multiple myeloma. Clin Cancer Res. 2016;22(22):5428-5433. doi: 10.1158/1078-0432.CCR-16-0866.
- Walker BA, Wardell CP, Melchor L, et al. Intraclonal heteroge- neity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood. 2012;120(5):1077-1086. doi: 10.1182/blood-2012-03-412981.
- Fonseca R, Bailey RJ, Ahmann GJ, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100(4):1417-1424.
- Lopez-Corral L, Sarasquete ME, Bea S, et al. SNP-based map- ping arrays reveal high genomic complexity in monoclonal gammo- pathies, from MGUS to myeloma status. Leukemia. 2012;26(12): 2521-2529. doi: 10.1038/leu.2012.128.
- Shah V, Sherborne AL, Walker BA, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular pro- files of 1905 trial patients [published online June 6, 2017]. Leukemia. doi: 10.1038/leu.2017.179.
- Walker BA, Wardell CP, Murison A, et al. APOBEC family mutational signatures are associated with poor prognosis transloca- tions in multiple myeloma. Nat Commun. 2015;6:6997. doi: 10.1038/ncomms7997.
- Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harous- seau JL, Bataille R; Intergroupe Francophone du Myelome. Rear- rangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001;98(10):3082-3086.
- Bolli N, Li Y, Sathiaseelan V, et al. A DNA target-enrichment approach to detect mutations, copy number changes and immu- noglobulin translocations in multiple myeloma. Blood Cancer J. 2016;6(9):e467. doi: 10.1038/bcj.2016.72.
- Lohr JG, Stojanov P, Carter SL, et al; Multiple Myeloma Research Consortium. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91-101. doi: 10.1016/j.ccr.2013.12.015.
- Walker BA, Boyle EM, Wardell CP, et al. Mutational spec- trum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33(33):3911-3920. doi: 10.1200/JCO.2014.59.1503.
- Chapman MA, Lawrence MS, Keats JJ, et al. Initial ge- nome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472. doi: 10.1038/nature09837.
- Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi: 10.1038/ncomms3997.
- Mikulasova A, Wardell CP, Murison A, et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica. 2017;102(9):1617-1625. doi: 10.3324/haematol.2017.163766.
- Dhodapkar MV. MGUS to myeloma: a mysterious gammopa- thy of underexplored significance. Blood. 2016;128(23):2599-2606.
- Mailankody S, Kazandjian D, Korde N, et al. Baseline mutation- al patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Advances. 2017;1(22):1911-1918. doi: 10.1182/bloodadvances.2017005934.
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415- 421. doi: 10.1038/nature12477.
- Catalogue of Somatic Mutations in Cancer. Signatures of muta- tional processes in human cancer. Sanger Institute website. cancer. sanger.ac.uk/cosmic/signatures. Published June 2017. Accessed November 2017.
- Liu Y, Chen C, Xu Z, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531(7595):471-475. doi: 10.1038/nature17157.
- Kalff A, Spencer A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: prognostic implications and current clinical strategies. Blood Cancer J. 2012;2:e89. doi: 10.1038/bcj.2012.37.
- Weinhold N, Ashby C, Rasche L, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood. 2016;128(13):1735-1744. doi: 10.1182/blood-2016-06-723007.
- Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067- 1076. doi: 10.1182/blood-2012-01-405985.
- Kortum KM, Mai EK, Hanafiah NH, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016;128(9):1226-1233. doi: 10.1182/blood-2016-02-698092.
- Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised Internation- al Staging System for Multiple Myeloma: a report from Internation- al Myeloma Working Group. J Clin Oncol. 2015;33(26):2863-2869. doi: 10.1200/JCO.2015.61.2267.
- Kumar S, Vij R, Kaufman JL, et al. Venetoclax monotherapy for relapsed/refractory multiple myeloma: safety and efficacy results from a phase I study. Blood. 2016;128(22):488-488.
- Moreau P, Chanan-Khan AA, Roberts AW, et al. Venetoclax combined with bortezomib and dexamethasone for patients with relapsed/refractory multiple myeloma. Blood. 2016;128(22):975-975.
- Morgan GJ, Jones JR. Integration of genomics into treatment: are we there yet? Am Soc Clin Oncol Educ Book. 2017;37:569-574. doi: 10.14694/EDBK_175166.
- Klco JM, Miller CA, Griffith M, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA. 2015;314(8):811-822. doi: 10.1001/jama.2015.9643.
- Miller C, Yesil J, Derome M, et al. A comparison of clinical FISH and sequencing based FISH estimates in multiple myeloma: an Mmrf Commpass Analysis. Blood. 2016;128(22):374.
- Lohr JG, Kim S, Gould J, et al. Genetic interrogation of circulat- ing multiple myeloma cells at single-cell resolution. Sci Transl Med. 2016;8(363):363ra147.
- Mithraprabhu S, Khong T, Ramachandran M, et al. Circulating tumour DNA analysis demonstrates spatial mutational heteroge- neity that coincides with disease relapse in myeloma. Leukemia. 2017;31(8):1695-1705. doi: 10.1038/leu.2016.366.





