Function of PI3K Pathway and Alterations in Breast Cancer
Class I phosphatidylinositol-3-kinases (PI3Ks) are heterodimers of a 110 KD catalytic subunit (p110α, p110β, p110γ or p110δ) and a regulatory subunit, which receive activation signals from receptor tyrosine kinases (RTKs), RAS, and G protein– coupled receptors (Figure 1).1 Activated PI3K catalyzes the conversion of phosphatidylinositol bisphosphate, PI(4,5)P2, to phosphatidylinositol triphosphate, PI(3,4,5)P3, which recruits phosphoinositide-dependent kinase-1 (PDK1) and protein kinase B, also known as AKT, leading to a cascade of signaling events that regulate cell survival, proliferation, metabolism, motility, and genomic stability.1 This pathway is also important in regulating tumor-associated immune response and angiogenesis.2

Genetic or epigenetic alterations in PI3K pathway components, including activating mutations in PIK3CA, the gene encoding the p110α catalytic subunit of PI3K, and AKT1, and loss-of-function mutations or epigenetic silencing of phosphatase and tensin homolog (PTEN), the negative regulator of the pathway, are commonly observed in cancer, leading to activation of PI3K pathway signaling. In estrogen receptor–positive (ER+) breast cancer, mutations in PIK3CA represent the most common genetic events, occurring at a frequency of 30% to 50%. Less commonly observed are mutations in PTEN (2% to 4%), AKT1 (2% to 3%), and phosphatidylinositol-3-kinase regulatory subunit alpha (PIK3R1: 1% to 2%).3,4 Similar findings were observed in HER2-positive breast cancer.4 In contrast, triple-negative breast cancer (TNBC) is associated with a lower incidence of PIK3CA mutations (<10%), but much higher frequency of loss of PTEN (about 30% to 50%).4 The frequent occurrence of PI3K pathway activation makes it an attractive therapeutic target in breast cancer.
Targeting the PI3K Pathway in ER+ Breast Cancer The importance of PIK3CA mutation in the etiology of ER+ breast cancer is supported by several lines of evidence. A majority of the mutations, including the 3 hotspot mutations E542K, E545K, and H1047R, are missense “activating” mutations that cluster in the evolutionarily conserved accessory domain and the kinase domain.5 The oncogenic property of the common PIK3CA mutations was demonstrated by their ability to induce cellular transformation and xenograft tumor formation when overexpressed in mammary epithelial cells.6,7 In preclinical studies, cancer cells carrying PIK3CA mutation depend on the alpha catalytic subunit of PI3K for cell growth.8 Although the presence of PIK3CA mutation in ER+ breast cancer has not been associated with de novo resistance to endocrine therapy,9 upregulation of PI3K pathway signaling has been observed in tumor cells grown under long-term estrogen deprivation in experimental models.10 In clinical samples, higher PI3K activity, based on the levels of phosphorylated forms of AKT, mammalian target of rapamycin (mTOR), glycogen synthase kinase 3 (GSK3), and the ribosomal protein S6 kinase (S6K), and loss of PTEN was associated with a lower ER level and with luminal B status.11 Importantly, a synthetic lethal interaction, or synergistic apoptotic induction, was observed between estrogen deprivation and inhibition of PI3K, either molecularly by knockdown of PIK3CA or pharmacologically with inhibitors of PI3K or AKT, in ER+ breast cancer cell lines.8,12 These studies provided the rationale for combining endocrine therapy and inhibitors of PI3K pathways in ER+ breast cancer.
Initial success from combing endocrine therapy with inhibitors against the PI3K pathway was demonstrated in clinical trials of rapamycin analogues for the treatment of advanced ER+ breast cancer resistant to an aromatase inhibitor (AI). These agents inhibit the activity of mTORC1 by interacting with FKBP12. In the TAMRAD trial, a randomized phase 2 trial of tamoxifen with or without everolimus in 111 postmenopausal women with AI-resistant, ER+, advanced breast cancer, the combination arm was associated with a significantly improved progression-free survival (PFS; 4.5 vs 8.6 months; hazard ratio [HR] = 0.54; P =.002) and overall survival (OS).13 Similarly, BOLERO-2, a phase 3 trial of exemestane in combination with either everolimus or placebo in postmenopausal women with advanced ER+, HER2-negative (HER2-) breast cancer resistant to letrozole or anastrozole, demonstrated a significant improvement in PFS (3.2 months in the placebo/exemestane arm vs 7.8 months in the everolimus/exemestane arm; HR = 0.45; P <.0001),14,15 leading to FDA approval of this combination in AI-resistant, advanced ER+ disease. However, the OS was no different in both arms (26.6 months in the placebo/exemestane arm vs 31 months in the everolimus/exemestane arm; HR = 0.89; P =.14).15 The OS data were disappointing; however, this may not be too surprising, as rapalogs lack the ability to inhibit mTORC2, leading to feedback upregulation of AKT activity and treatment failure.16 Nonetheless, the improvement in PFS is meaningful, and everolimus remains a treatment option for the AI-resistant population.
Everolimus was also combined with fulvestrant in a singlearm, phase 2 trial in AI-resistant, metastatic, ER+ breast cancer.17 Among the 31 evaluable patients, the objective response rate was 13% and the clinical benefit rate was 49%, with median time to progression (TTP) of 7.4 months, suggesting clinical efficacy. However, a randomized trial is required to define the activity of this combination in AI-resistant, ER+ breast cancer.
A number of agents that target the PI3K pathway (Table 1), including mTOR kinase inhibitors, direct PI3K or AKT inhibitors, and dual inhibitors of PI3K and mTOR, are in clinical development and have the potential to more effectively inhibit the PI3K pathway than rapamycin analogues.18 The mTOR kinase inhibitors and AKT inhibitors are in early phases of clinical trial development, while PI3K inhibitors have advanced to phase 3 trials and have shown promise in early-phase trials for ER+ disease.19
PI3K inhibitors are classified into pan-PI3K inhibitors and isoform-specific inhibitors, depending on their specificities to the 4 isoforms of the p110 catalytic domain, p110α, p110β, p110γ, and p110δ. Isoform-specific inhibitors may have the advantage of more effective isoform inhibition at tolerable doses than pan-PI3K inhibitors. However, patient selection may be particularly important for these agents, as cancers may rely on different p110 isoforms for cell growth. For example, p110α is critical for PIK3CA-mutant breast cancers, while p110β appears to be particularly important for those with loss of PTEN. Therefore, p110α-specific inhibitors may not be effective in tumors deficient of PTEN.
Early trials of PI3K inhibitors have shown promising activity in ER+ breast cancer. In a phase 1b study of buparlisib, a pan-PI3K inhibitor, plus letrozole in patients with metastatic ER+ breast cancer refractory to endocrine therapy, 16 of 51 patients (31%) enrolled in the study derived clinical benefit (lack of disease progression, ≥6 months).19 Buparlisib has also been combined with fulvestrant, with activity observed in a phase 1 trial that enrolled patients with metastatic ER+ breast cancer.20 The combination of buparlisib and fulvestrant is being evaluated in phase 3 trials, including BELLE-2 (NCT01610284) for AI-resistant, metastatic, ER+ breast cancer, and BELLE-3(NCT01633060) for AI-resistant, metastatic, ER+ breast cancer that has progressed on or after a mTOR inhibitor.
Pictilisib is the first pan-PI3K inhibitor for which results of randomized trials in ER+ breast cancer have been reported.21,22 OPPORTUNE is a preoperative window study that randomized 75 postmenopausal patients with newly diagnosed, operable, ER+, HER2-negative breast cancer at a 2:1 ratio to receive 2-week preoperative treatment with anastrozole plus pictilisib (n = 49, 44 evaluable) or anastrozole alone (n = 26, all evaluable), with the primary endpoint of inhibition of tumor-cell proliferation, as measured by change in Ki67 expression at surgery following 2-week treatment.21 A higher degree of Ki67 suppression was observed with the combination therapy (83.8%) compared with anastrozole alone (66%; P =.004), indicating a superior efficacy of the combination arm.21 The FERGI phase 2 study of pictilisib plus fulvestrant versus fulvestrant plus placebo in patients with AI-resistant, ER+, advanced or metastatic breast cancer was reported at the 2014 San Antonio Breast Cancer Symposium (SABCS). The median PFS was 5.1 months (fulvestrant + placebo arm) vs 6.6 months (fulvestrant + pictilisib arm; P =.0959), which did not differ statistically. However, in the progesterone receptor–positive (PR+) subgroup, the addition of pictilisib to fulvestrant resulted in an improvement of PFS from 3.7 months to 7.4 months (HR = 0.440; 95% CI, 0.281-0.689).22 In this trial, dose reductions of pictilisib due to skin and gastrointestinal (GI) toxicities were frequent, and few patients experienced the ontarget side effect of PI3K inhibition with hyperglycemia, arguing that perhaps the dose of pictilisib might not have been optimal. We await the release of the BELLE2 data, which is anticipatedearly 2015, and data from other trials of isoform-selective PI3K inhibitors to define the role of various PI3K inhibitors in the treatment of ER+ breast cancer.
Targeting the PI3K Pathway in HER2-Positive Breast Cancer
It is well established that HER2-amplified tumors show significant dependence on the PI3K pathway, and the antitumor effect of HER2-targeted agents is at least in part mediated by inhibition of PI3K pathway activity.23 Preclinical data demonstrated that the presence of PIK3CA mutations in HER2-positive breast cancer uncouples HER2 and PI3K signaling, rendering tumor cells resistant to HER2-targeted agents such as trastuzumab, while dual targeting of the HER2 and PI3K pathways was effective in overcoming trastuzumab resistance.23

However, addition of the mTOR inhibitor everolimus to trastuzumab-containing chemotherapy regimens for the treatment of metastatic HER2+ breast cancer has led to disappointing results in clinical trials. In the setting of the treatment of trastuzumaband taxane-resistant metastatic HER2+ breast cancer, addition of everolimus to the combination of trastuzumab and vinorelbine led to a statistically significant but small improvement in median TTP (7.0 months with everolimus vs 5.8 months without everolimus; P <.01) in the randomized phase 3 BOLERO-3 trial, reported at the 2014 American Society of Clinical Oncology Annual Meeting.24 In the first-line metastatic setting, addition of everolimus to the combination of trastuzumab and paclitaxel showed no improvement in PFS (14.95 months with everolimus vs 14.49 months without everolimus) in the phase 3 BOLERO-1 trial, reported at the 2014 SABCS.25 However, in both trials, the ER-negative (ER-) population derived more benefit, arguing the potential role of everolimus in the ER population.
Clinical trials of other PI3K pathway inhibitors are under way for the treatment of HER2+ breast cancer.26 In a phase 1b trial of buparlisib plus trastuzumab in patients with HER2+ advanced or metastatic breast cancer that progressed on trastuzumab-based therapy, the combination was well tolerated; at the recommended phase 2 dosage, there were 2 (17%) partial responses and 7 (58%) patients had stable disease (≥6 weeks), suggesting clinical activity.27 A phase 2 study of this combination is ongoing to define the role of PI3K inhibitors in the treatment of HER2+ breast cancer.
Targeting the PI3K Pathway in TNBC
The potential role of the PI3K pathway in the pathogenesis of TNBC is supported by the frequent detection of PTEN loss and the evidence of activated PI3K pathway signaling in this subtype of breast cancer in an analysis of The Cancer Genome Atlas (TCGA) samples.4 In preclinical studies, inhibitors against mTOR and AKT induced growth arrest of patient-derived xenograft (PDX) models of TNBC.28 In addition, in the phase 1 study of single-agent BKM120, a pan-PI3K inhibitor, a partial response was observed in a patient with TNBC.29 Further preclinical and clinical studies that define the role of PI3K in TNBC are under way. However, frequent mutations or copy number changes in genes important for cell cycle arrest or apoptosis, such as RB, MYC, and TP53, likely limit the antitumor activity of PI3K pathway inhibitors in this tumor type. Rational combination therapies likely are needed. Of particular interest is the discovery that the PI3K pathway plays an important role in maintaining the genomic stability, and that inhibitors of the PI3K pathway may increase DNA damage and sensitize TNBC to inhibitors of poly ADP ribose polymerase (PARP).30,31 The combination of inhibitors against PI3K and PARP is being tested in clinical trials for the treatment of advanced TNBC.
Adverse Events of PI3K Pathway Inhibitors
In general, PI3K pathway inhibitors including mTOR, AKT, and PI3K inhibitors have been shown to be well tolerated. Common adverse events (AEs) associated with everolimus include stomatitis, rash, fatigue, GI side effects, pneumonitis, and hyperglycemia. However, most of these AEs are grade 1 and 2. Among these AEs, stomatitis is the most common cause for dose interruption/dosage adjustment. In the BOLERO 2 trial, stomatitis occurred in 59% of patients (8% grade 3).32 Everolimus-induced stomatitis often presents as aphthous-like ulcers that develop acutely within the first 2 weeks of treatment and quickly resolves with dose interruption. Prophylactic measures to promote good oral hygiene are recommended, including the use of a soft toothbrush that is changed on a regular basis; daily flossing; frequent rinsing with bland rinses such as sterile water, normal saline, or sodium bicarbonate. Avoidance of acidic, spicy, and hard or crunchy foods and alcoholic mouthwash are highly recommended. In addition, close follow-up of patients after initiation of treatment with dose interruption and dosage reduction after resolution is important.32 In contrast to stomatitis associated with chemotherapy or radiation therapy, everolimus-induced stomatitis has an inflammatory component. The use of topical high-potency corticosteroids (eg, dexamethasone 0.1 mg/mL; clobetasol gel 0.05 %), topical nonsteroidal anti-inflammatories (eg, amlexanox 5 % oral paste), and topical anesthetics (“miracle” or “magic” mouthwashes typically containing lidocaine viscous, diphenhydramine, and an antacid such as aluminum hydroxide or magnesium hydroxide) are recommended.32,33
In clinical trials of PI3K inhibitors, common AEs included hyperglycemia, GI toxicity, fatigue, transaminitis, and skin rash.19,22 Mood disorder was also seen in trials of buparlisib. Hyperglycemia is an on-target side effect of PI3K inhibition, as a result of more sustained inhibition of p110a, and is often manageable with metformin.19,21,29 It remains to be determined whether isoform-specific inhibitors are more advantageous in widening the therapeutic window of PI3K inhibitors.
Predictors of Response
The identification of genetic predictors of response to PI3K pathway inhibitors has been complicated by the presence of a multitude of genetic or epigenetic alterations of the pathway components and the lack of relevant tumor specimens.34 Singlegene alterations such as PIK3CA mutation have not been predictive of treatment response in studies of everolimus and pan-PI3K inhibitors.19 In an effort to identify the target population of everolimus, archival tumor specimens from patients enrolled in the BOLERO-2 trial were subjected to targeted next-generation sequencing (NGS) of 182 cancer-related genes. No correlation with PFS was observed with each of the 9 genes with a mutation rate >10% (eg, PIK3CA, FGFR1, and CCND1), or when less frequently mutated genes (eg, PTEN, AKT1) were included in their respective pathways.34 However, most of the archival tumor specimens analyzed were obtained from the primary rather than the metastatic disease. Hypothetically, RNA or protein signatures that measure the signaling output of the PI3K pathway could be more informative. However, there is no established clinical assay for this approach at this time. Limited protein markers of PI3K pathway activity were evaluated in the translational study of TAMRAD, in which p4EBP1, LKB1, or PI3K protein expression were found to be predictive of everolimus benefit.35 Larger studies are needed to confirm the results. At this time, there are no established biomarkers available for patient selection.
Similarly, in clinical trials of pan-PI3K inhibitors, PIK3CA mutation or PTEN status has not been associated with the antitumor response in clinical trials.19,21,22 Interestingly, in the neoadjuvant pictilisib trial, luminal B cancers derived more benefit from the addition of pictilisib in Ki67 suppression.21 An exploratory subgroup analysis in the FERGI trial demonstrated that addition of pictilisib to fulvestrant significantly improved PFS in the subgroup of patients with PR+ tumors.22 However, these results need to be validated in other trials. Perhaps isoformspecific inhibitors are more likely to function in genetically defined populations based on their mechanisms of action.
Combination Treatment Strategies
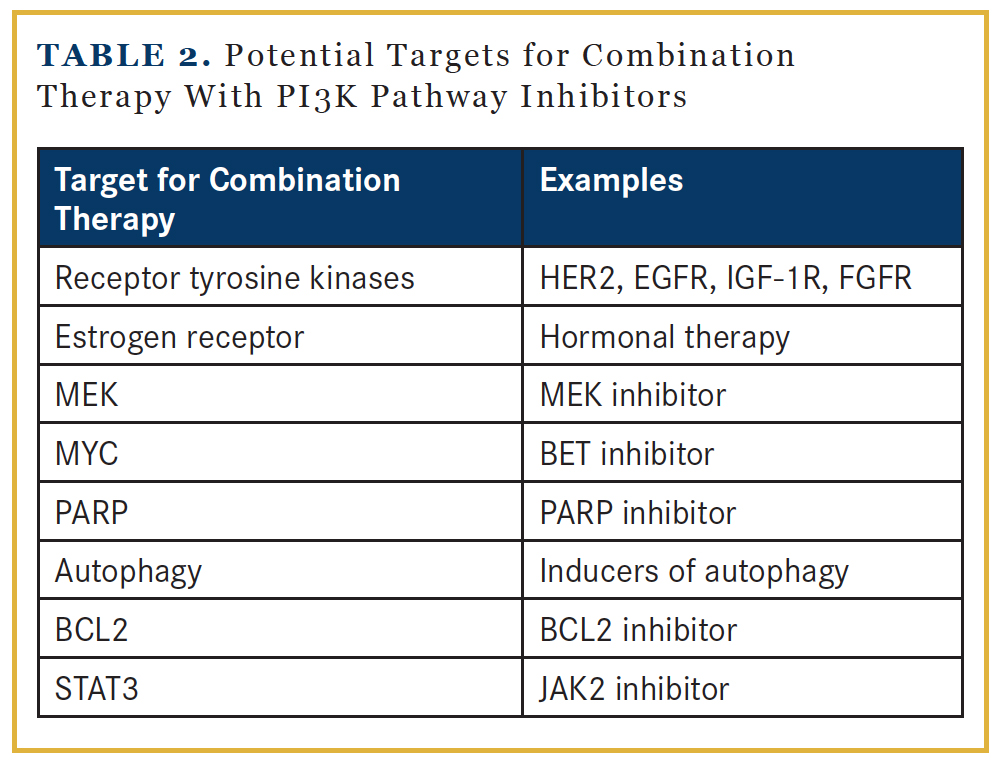
The antitumor activity of single-agent PI3K pathway inhibitors is likely limited due to the presence of feedback loops within the PI3K pathway and crosstalk between PI3K pathway and signaling pathways. Successful tumor control likely demands combination therapy approaches. Based on preclinical data, potential candidate partners include inhibitors against RTKs, MEK, MYC, PARP, or the STAT3 pathway, and strategies to enhance autophagy and apoptosis (Table 2).18 Inhibition of the PI3K pathway leads to FOXO nuclear accumulation that upregulates the expression of RTK.36,37 Examples of ongoing clinical trials that target both the RTK and PI3K pathways include HER2-targeted agents in combination with PI3K inhibitors in HER2+ breast cancer.38 However, combining RTK inhibitors with PI3K pathway inhibitors may be challenging, as several RTKs could be upregulated. An alternative approach is to combine PI3K pathway inhibitors with inhibitors of the RAS/ RAF/MEK/ERK cascade, as both mediate RTK signaling and promote cell survival and proliferation. The extensive crosstalk between these 2 pathways has been demonstrated in preclinical studies. PI3K pathway inhibition activates ERK,39 while MEK inhibition increases AKT activity.40 Clinical trials are ongoing to test the tolerability of this strategy.41 Data indicated increased toxicity with dual blockade of the PI3K and MEK pathways, but treatment could be particularly effective in tumors with genetic alterations in both pathways.42 Inhibitors of bromodomain and extraterminal domain (BET), which regulate the transcription level of MYC, may be reasonable approaches in tumors with MYC overexpression, as it has been shown to confer resistance to PI3K pathway inhibitors.43 BCL2 family proteins produce an antiapoptotic effect by maintaining mitochondrial integrity. Preclinical evidence supports the use of BCL2 antagonists, to prime for mitochondrial death, in combination with PI3K pathway inhibitors.44
Conclusion
The PI3K pathway is an important therapeutic target in breast cancer. In ER+ breast cancer, initial success has been observed with mTOR inhibitors. In addition, clinical trials of mTOR kinase inhibitors and direct inhibitors of PI3K and AKT, which potentially inhibit the pathway more effectively, are ongoing. However, as single agents, the antitumor activity of PI3K pathway inhibitors is likely limited as a result of feedback regulation and crosstalk with RTK and other signaling pathways. Strategies that combine PI3K pathway inhibitors with inhibitors against RTKs, or inhibitors against MEK, MYC, PARP, or STAT3 pathways, or agents that activate autophagy and apoptosis machineries, are being explored. In addition, there is continued effort to identify resistance mechanisms and predictors of therapeutic response.
Affiliation: Cynthia X. Ma, MD, PhD, is associate professor of Medicine, Division of Oncology, Department of Medicine, Washington University School of Medicine, St. Louis, MO.
Disclosure: Dr Ma reports receiving research funding from Novartis, Pfizer, and Puma Biotechnology.
Address correspondence to: Cynthia X. Ma, MD, PhD, Associate Professor of Medicine, Division of Oncology, Department of Medicine, Campus Box 8056, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110; phone: (314) 362-9383; fax: (314) 362-7086; email: [email protected].
References
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627-644.
- Hirsch E, Ciraolo E, Franco I, et al. PI3K in cancerstroma interactions: bad in seed and ugly in soil. Oncogene. 2014;33(24):3083-3090.
- Ma CX, Crowder RJ, Ellis MJ. Importance of PI3-kinase pathway in response/resistance to aromatase inhibitors. Steroids. 2011;76(8):750-752.
- Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61-70.
- Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65(7):2554-2559.
- Zhang H, Liu G, Dziubinski M, et al. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat 2008;112(2):217-227.
- Zhao JJ. The oncogenic properties of mutant p110[agr] and p110[bgr] phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci. 2005;102:18443-18448.
- Crowder RJ, Phommaly C, Tao Y, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor positive breast cancer. Cancer Res. 2009;69(9):3955-3962.
- Ellis MJ, Lin L, Crowder R, et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res Treat. 2010;119(2):379-390.
- Miller TW, Hennessy BT, Gonzalez-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406-2413.
- Creighton CJ, Fu X, Hennessy BT, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12(3):R40.
- Sanchez CG, Ma CX, Crowder RJ, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13(2):R21.
- Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30(22):2718-2724.
- Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520-529.
- Piccart M, Hortobagyi GN, Campone M, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol. 2014;25(12):2357-2362.Cancer Res. 2006;66(3):1500-1508.
- O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500-1508.
- Massarweh S, Romond E, Black E, et al. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res Treat. 2014;143(2):325-332.
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature Rev Drug Discov. 2014;13(2):140-156.
- Mayer IA, Abramson VG, Isakoff SJ, et al. Stand Up to Cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2014;32(12):1202-1209.
- Ma CX, Luo J, Naughton M, et al. A phase I study of BKM120 and fulvestrant in postmenopausal women with estrogen receptor positive metastatic breast cancer. Presented at: the 37th Annual San Antonio Breast Cancer Symposium; December 9-13, 2014; San Antonio, TX. Abstract PD5-6.
- Schmid P, Pinder SE, Wheatley D, et al. Preoperative window of opportunity study of the PI3K inhibitor pictilisib (GDC-0941) plus anastrozole vs anastrozole alone in patients with ER+, HER2-negative operable breast cancer (OPPORTUNE study). Presented at: the 37th Annual San Antonio Breast Cancer Symposium; December 9-13, 2014; San Antonio, TX. Abstract S2-03.
- Krop I, Johnston S, Mayer IA, et al. The FERGI phase II study of the PI3K inhibitor pictilisib (GDC-0941) plus fulvestrant vs fulvestrant plus placebo in patients with ER+, aromatase inhibitor (AI)-resistant advanced or metastatic breast cancer – part I results. Presented at: the 37th Annual San Antonio Breast Cancer Symposium; December 9-13, 2014; San Antonio, TX. Abstract S2-02.
- Rexer BN, Chanthaphaychith S, Dahlman K, Arteaga CL. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 2014;16(1):R9.
- O’Regan R, Ozguroglu M, Andre F, et al. Phase III, randomized, double-blind, placebo-controlled multicenter trial of daily everolimus plus weekly trastuzumab and vinorelbine in trastuzumab-resistant, advanced breast cancer (BOLERO-3). J Clin Oncol. 2013;31(suppl; abstr 505).
- Hurvitz SA, Andre F, Jiang Z, et al. Phase 3, randomized, double-blind, placebo-controlled multicenter trial of daily everolimus plus weekly trastuzumab and paclitaxel as firstline therapy in women with HER2+ advanced breast cancer: BOLERO-1. Presented at: the 37th Annual San Antonio Breast Cancer Symposium; December 9-13, 2014; San Antonio, TX. Abstract S6-01.
- Rexer BN, Arteaga CL. Optimal targeting of HER2-PI3K signaling in breast cancer: mechanistic insights and clinical implications. Cancer Res. 2013;73(13):3817-3820.
- Saura C, Bendell J, Jerusalem G, et al. Phase Ib study of buparlisib plus trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on trastuzumab-based therapy. Clin Cancer Res. 2014;20(7):1935-1945.
- Xu S, Li S, Guo Z, et al. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patientderived xenograft models. Mol Cancer Ther. 2013;12(8):1665-1675.
- Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282-290.
- Ibrahim YH, Garcia-Garcia C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036-1047.
- Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048-1063.
- Peterson ME. Management of adverse events in patients with hormone receptor-positive breast cancer treated with everolimus: observations from a phase III clinical trial. Support Care Cancer. 2013;21(8):2341-2349.
- Porta C, Osanto S, Ravaud A, et al. Management of adverse events associated with the use of everolimus in patients with advanced renal cell carcinoma. Eur J Cancer. 2011;47(9):1287-1298.
- Hortobagyi GN, Piccart-Gebhart MJ, Rugo HS, et al. Correlation of molecular alterations with efficacy of everolimus in hormone receptor–positive, HER2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2013;31(suppl; abstr LBA509).
- Treilleux I, Arnedos M, Cropet C, et al. Predictive markers of everolimus efficacy in hormone receptor positive (HR+) metastatic breast cancer (MBC): final results of the TAMRAD trial translational study. J Clin Oncol. 2013;31(suppl; abstr 510).
- Chakrabarty A, Sanchez V, Kuba MG, et al. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci. 2012;109(8):2718-2723.
- Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1(3):248-259.
- Singh JC, Jhaveri K, Esteva FJ. HER2-positive advanced breast cancer: optimizing patient outcomes and opportunities for drug development. Br J Cancer. 2014;111(10):1888-1898.
- Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065-3074.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50(1):43-55.
- Saini KS, Loi S, de Azambuja E, et al. Targeting the PI3K/ AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39(8):935-946.
- Shimizu T, Tolcher AW, Papadopoulos KP, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316-2325.
- Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYCeukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci. 2011;108(37):E699-708.
- Rahmani M, Aust MM, Attkisson E, et al. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013;73(4):1340-1351.





