Introduction
Metastasis is responsible for more than 90% of cancer-related deaths. Prostate cancer is no exception, with approximately 26,120 men expected to succumb to the disease due to complications of metastasis in 2016.1 Of these, it is expected that more than 90% will have evidence of skeletal lesions.2 The median survival time for patients with active metastatic castration-resistant prostate cancer (mCRPC) is approximately 3 years. Understanding how metastatic prostate cancer cells grow and interact with the surrounding tumor microenvironment can identify key circuits driving the progression of the disease.3 Research in this area has revealed targets for therapeutic intervention, the translation of which should enhance the overall survival (OS) of patients with mCRPC.
Androgen Deprivation Therapy (ADT) for Bone mCRPC
The National Comprehensive Cancer Network suggested guidelines4 for the treatment of men given a diagnosis with bone mCRPC are immunotherapy (sipuleucel-T) followed by androgen deprivation therapy (ADT; abiraterone acetate and enzalutamide), chemotherapy (docetaxel with prednisone), radiopharmaceutical therapy (radium 223), suggestion of a clinical trial, or a potential secondary hormone therapy such as ketaconozole.
The upregulation of pathways involved in androgen synthesis or mutations/amplification in the androgen receptor (AR) itself allows cancer cells to continue feeding on androgens despite the systemic depletion of the ligand. Further, androgen interaction with AR-expressing bone-building osteoblasts promotes differentiation and bone formation.5,6 Given the reliance of mCRPC cells on androgen for growth in bone, inhibitors that block androgen synthesis or the activity of mutant AR remain an intense area of investigation and clinical trial activity. For example, CYP17A1 is an important enzyme used by CRPC cells for the de novo synthesis of androgens, and this discovery led to the genesis of abiraterone, a small molecule inhibitor of CYP17A1 activity.7 Abiraterone given in combination with prednisone, a corticosteroid, was first shown to increase the median OS by 4.6 months, compared with placebo plus prednisone, in mCRPC patients who had previously received docetaxel.8 Median OS was increased to three years in chemotherapy-naïve patients, compared with placebo.9 Additionally, abiraterone was shown to also significantly delay the time to first skeletal-related event (SRE).10,11
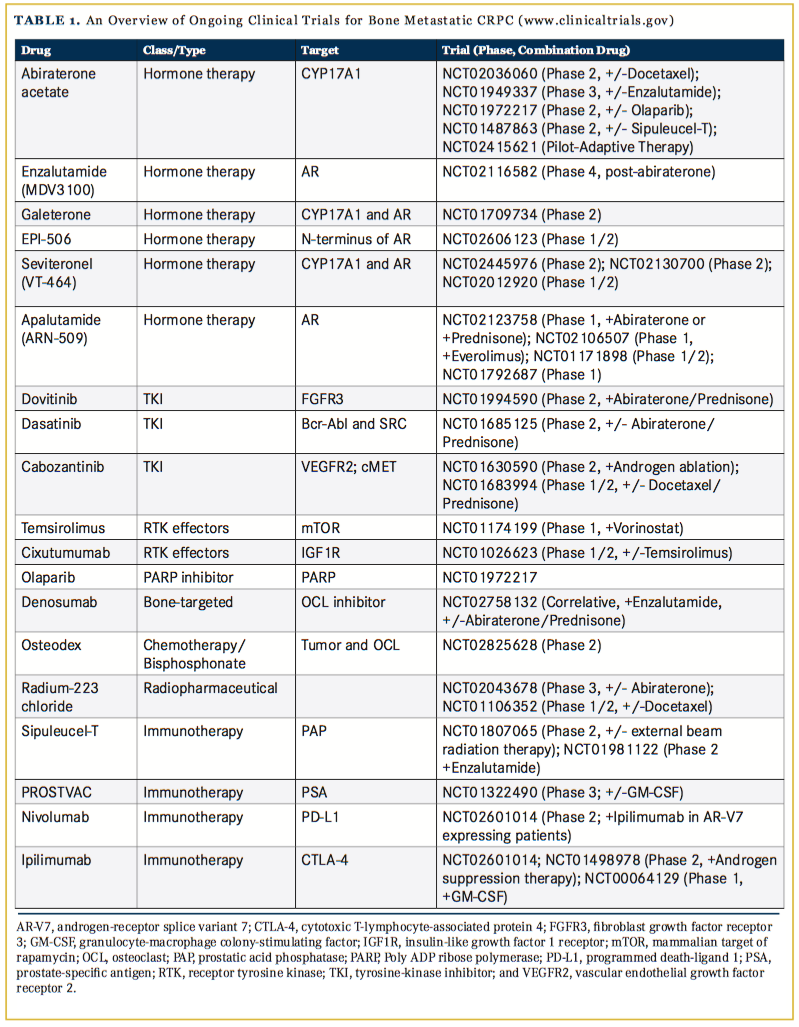
Enzalutamide, an AR antagonist, was first shown to increase median OS by 4.8 months in mCRPC patients who had previously received docetaxel, compared with the placebo group.12 The time-to- disease progression, measured by prostate-specific antigen (PSA) levels, was increased by 5.3 months and radiographic progression-free survival (PFS) increased by 5.4 months, compared with placebo. In a phase III randomized trial, enzalutamide increased the time to the first occurrence of an SRE, suggesting an impact on disease progression in bone.13 Given the success of these ADTs as single agents, they are now being investigated for their efficacy together or when combined with other therapies (Table).
Building on this approach, galeterone, a novel dual small molecule inhibitor of CYP17A1 and AR was compared with enzalutamide alone in clinical trials (ARMOR3-SV). The major endpoint for mCRPC patients was radiographic PFS but in July 2016, the trial was halted due to predicted failure to meet this goal.14 The drug, however, remains in phase II clinical trials examining safety and response of the patients that have progressive CRPC but have failed oral therapy (ARMOR2; NCT01709734). Other AR and CYP17A1 targeted inhibitors, such as apalutamide and seviteronel, remain the focus of phase I and II clinical trials (Table 1).
While results for these inhibitors have been encouraging, a caveat has been the emergence of AR variants (AR-V) that, in some instances, lack ligand-binding domains but still drive the expression of AR-related genes. Recently, AR-V7 has been linked to acquired resistance to enzalutamide and abiraterone.15 EPI-506 is a novel small molecule inhibitor that binds to the N-terminal domain of AR and therefore could be an effective treatment for mCRPC patients who have developed resistance to enzalutamide.16 The long-term safety of EPI-506 is currently being studied (NCT02606123).
Targeting Bone mCRPC From the Outside In
Although the emphasis has remained on ADT, understanding ligands, receptors, and signaling pathways that control CRPC has revealed critical circuits controlling cancer well in survival and growth. The mutation/amplification/upregulation of several receptor tyrosine kinases (RTKs) have been implicated in the development, growth, and progression of prostate cancer and are the focus of clin- ical trials.17 For example, dovitinib, a tyrosine kinase inhibitor (TKI) that binds fibroblast growth factor receptor 3 (FGFR3), is currently under investigation for efficacy in combination with abiraterone (NCT01994590), after being previously shown to improve bone scans and reduce SREs in 6 of 23 patients in a proof-of-principle study.18 FGF signaling in bone stromal cells is an important regulator of bone formation, and it is possible dovitinib can impact prostate cancer cell growth and osteoblast behavior.19
Overall, TKI trial results for the treatment of mCRPC have been varied. Dasatinib, an inhibitor of multiple TKIs including SRC family kinases, reduced disease progression in 57% and bone lesions in 30% of mCRPC patients in a phase I trial.20 However, in a recent phase 3 trial, the combination of dasatinib and docetaxel failed to provide a survival advantage compared with docetaxel and placebo.21 Interestingly, dasatinib has been shown to induce differentiation of mesenchymal stromal cells in bone-forming osteoblasts, which may exacerbate prostate tumor-induced osteogenesis.22 Combination therapy with an anti-androgen may circumvent this possibility. To that end, a combinational study of dasatinib and abiraterone/prednisone prior to chemotherapy is currently being investigated for impact on PFS as a primary outcome (NCT01685125).
Constitutive activation of multiple signaling pathways via different RTKs can provide a significant survival advantage for tumors; thus dual targeting of TKIs may be beneficial for impacting tumor growth. Cabozantinib, for example, is a dual TKI of VEGFR2, the receptor for angiogenic factor vascular endothelial growth factor (VEGF), and c-MET, a receptor for hepatocyte growth factor (HGF). In a phase II randomized trial, daily administration of cabozantinib improved bone scans in 68% of mCRPC patients (with complete resolution in 12%), reduced soft tissue lesions, and improved PFS.23,24 However, cabozantinib failed to reach the primary endpoint of increasing OS, compared with prednisone alone, in a phase III randomized trial of mCRPC patients who had previously received docetaxel and abiraterone.25 Trials examining cabozantinib in combination with androgen ablation (NCT01630590) or chemotherapies such as docetaxel (NCT01683994) are ongoing and recruiting. RTKs mediate their effects via cell signaling circuitry and inhibitors of RTK effectors — such as mTOR for example — are also being explored clinically (NCT01174199 and NCT01026623).
Under selective pressures induced by therapeutic regimens, prostate cancer cells often acquire resistance to programmed cell death. For example, upregulation of DNA repair mechanisms is a common way for prostate cancer cells to avoid apoptosis induced by environmental stress.26,27 Currently, inhibitors of poly ADP ribose polymerases (PARPs) that repair DNA “nicks” are being investigated. PARP-1 is a nuclear enzyme that detects single- and double-strand DNA breaks and initiates repair mechanisms. Further, PARP-1 can bind and regulate AR transcriptional function. PARP-1 has also been shown to play a critical role in mesenchymal stem cell--driven osteogenesis, making it a promising target for treating bone mCRPC.21,28 Olaparib (Lynparza), a PARP-1 inhibitor, was included in a phase II trial of 50 mCRPC patients, 16 of which had mutational defects in DNA-repair genes,29 in bone and visceral organ metastasis biopsies measured before and after treatment. Olaparib produced a PSA response (decline of 50% or more) in 22% of the patients and reduced the numbers of circulating tumor cells in 29%. Eighty-eight percent of patients with defects in DNA-repair genes (including BRCA1, ATM, CHEK2, and HDAC) showed a positive response to olaparib, suggesting that mutations in DNA repair genes may serve as a biomarker for mCRPC response to PARP inhibition. A current trial is examining the efficacy, safety, and tolerance of olaparib given in combination with abiraterone and will be compared with placebo with abiraterone (NCT01972217).
Bone Microenvironment Targeted Therapies for mCRPC Treatment
The surrounding bone microenvironment is a key driver of CRPC growth, and as such, presents therapeutic opportunities.3 Although a hallmark of mCRPC is bone formation, the lesions also contain areas of extensive osteolysis and osteoclast activity. The monoclonal antibody denosumab binds to the receptor-activator of nuclear factor kappa B-ligand (RANKL), a key regulator of osteoclast formation. By preventing interaction with its cognate receptor RANK, denosumab effectively inhibits osteoclast formation and activation. Denosumab has been proven to significantly increase the median time to 1 SRE by 18% (20.7 months vs 17.1 months) compared with bisphosphonates.30 Despite these results, no impact on OS of the patients was noted compared with the control arm. Because denosumab is well tolerated, it is currently being explored in combination with other therapies such as abiraterone (NCT02758132).
Another class of bone-targeted inhibitors commonly used for the treatment of mCRPC is bisphosphonates. Bisphosphonates specifically target normal and pathological bone formation by binding to calcium in newly-formed bone, and upon resorption, they are taken up by osteoclasts, inducing their apoptosis.31 Compared with placebo, bisphosphonates such as zoledronate significantly increased median time to a SRE (488 days vs 321 days for placebo treatment) and reduced the frequency of SREs (39% vs 49%). Similar to denosumab, zoledronate does not enhance OS of men with mCRPC.32,33 Bisphosphonates are also well tolerated in patients and thus provide an advantageous strategy for delivering therapies to the bone tissue and, specifically, areas undergoing remodeling. Guanidine, for example, is a potent chemotherapy but noted side effects make applying the treatment to patients difficult.34 Osteodex is a novel therapy that grafts guanidine onto a bisphosphonic foundation with the goal of specifically targeting bone metastases and avoiding dose-limiting toxicities via bone-specific delivery of guanidine. Osteodex is currently in phase II trials that are investigating time to SRE (NCT02825628). A recent breakthrough has been the FDA approval of radium-223 dichloride. The radium isotope is similar in nature to calcium and is preferentially absorbed by bone tissue where it emits high-energy alpha particles, killing cancer cells within a short range (less than 100 mcm). Radium-223 was found to improve median OS by 3.6 months compared with placebo.35 Because of its success, clinical trials are investigating the efficacy of radium-223 with other therapies such as abiraterone (NCT02043678) and docetaxel (NCT01106352).
Significant advances have been made in the past decade with the development of immune-targeted therapies aimed at activating anti-tumor immunity and inhibiting pro-tumoral immunity. The immunostimulant sipuleucel-T and immune vaccine, PROSTVAC, activate the immune system against 2 well- defined prostate antigens, prostate acid phosphatase (PAP) and PSA, respectively. Sipuleucel-T is a personalized treatment involving ex vivo culture of patient-derived antigen-presenting cells (APCs) with a fusion protein (PA2024) of recombinant PAP and granulocyte-macrophage colony-stimulating factor (GM-CSF), an immune stimulating factor. APC-expressing PA2024 cells are then transfused back into the patient where they induce immune activation against cancer-derived PAP. Compared with placebo, sipuleucel-T proved to be most beneficial for mCRPC patients with low disease burden, improving median OS by 4 months and 3-year survival, but has not been as successful for patients with more advanced disease (>20 detected bone lesions), demonstrating a need for greater understanding of mCRPC immunogenicity in bone.36-38
PROSTVAC utilizes 2 recombinant poxviruses: vaccinia (PROS- TVAC-V), which primes the immune system; and fowlpox (PROST- VAC-F), an immune system booster. Each vector has been transduced to express 4 human genes: PSA and 3 costimulatory molecules that enhance T-cell activation (leukocyte function-associated antigen-3, [LFA3]; intercellular adhesion molecule-1, [ICAM1]; and B7-1).39 In a phase II trial, PROSTVAC significantly improved median OS in mCRPC patients.40 These findings contributed to the initiation of an ongoing phase III trial investigating the impact of PROSTVAC alone or in combination with GM-CSF on overall survival in asymptomatic mCRPC patients (NCT01322490).
A new wave of immunotherapies has arisen in recent years that specifically targets checkpoint inhibition, a mechanism of tumor immune evasion that prevents cytotoxic T-cell lymphocyte (CTL) activation. Although the percentage of T cells in the bone marrow is relatively low, CD4+ and CD8+ CTLs have been shown to exert anti-tumor effects in bone metastases of other cancers, such as breast and melanoma.41-43 Ipilimumab, a monoclonal antibody against receptor cytotoxic T-lymphocyte antigen-4 (CTLA-4), a negative regulator of T-cell activation, inhibits regulatory T-cell function and activates cytotoxic T-cells. Although ipilimumab has been highly successful for the treatment of metastatic melanoma,44,45 it has not proved efficacious for the treatment of mCRPC. In a recent phase III trial, ipilimumab failed to improve overall survival in comparison to placebo, yet reduced PSA, and improved 3-month progression-free survival.42
Several studies have demonstrated adverse effects that ended initial clinical trials but of note, a single patient had a dramatic response with a reduction in the number of bone lesions and disease free survival at 6 years.46 Defining markers predictive of patient response to checkpoint inhibitors will be critical for their clinical application. Nevertheless, clinical trials are investigating the combination of ipilimumab with ADT on disease progression (NCT01498978) and the safety of using ipilimumab in combination with GM-CSF (NCT00064129). Another checkpoint inhibitor drug, nivolumab, a monoclonal antibody that targets PD-1/PD-L1, has shown little promise, as there has been no clear indication that CRPC tumors express PD-L1.47 Targeting T-cell activation through 2 different mechanisms, APC-mediated activation and immune checkpoint inhibition, such as combinational PROSTVAC with ipilumimab treatment, may enhance drug efficacy over the individual compounds. Clinical trials are studying the efficacy of the checkpoint inhibitors combined (NCT02601014) or when added to ADT (NCT01498978).
Upcoming Opportunities and Threats for Bone mCRPC Treatment
Significant progress has been made in the development of therapies that target mCRPC growth in the bone microenvironment. Moving forward, the upfront application of therapies in combination — such as ADT with radium-223 — for example, may prove more effective than sequential treatments in extending OS. Molecular profiling of individual mCRPC patients and the identification of response predictors will clearly be beneficial for the smart application of targeted therapies, such as TKIs and immune check point inhibitors, in order to achieve maximal responses. A major challenge for the medical oncologists is mCRPC heterogeneity and the emergence of resistant disease.48,49 Adaptive therapy aims to prevent the emergence of resistant subpopulations by maintaining therapy-sensitive populations.50 The application of therapies, as needed, to stabilize disease progression, rather than continuously, is currently being explored in the clinic for abiraterone in mCRPC (NCT02415621). Further, novel computation- al modeling approaches to define optimal therapeutic strategies for heterogeneous bone metastatic prostate cancer are under investigation.51-53
In conclusion, a greater understanding of the molecular underpinnings of bone metastasis has contributed to an expansion of potential therapies for mCRPC. Defining the optimal sequence and combinations needed for these therapies, identifying key characteristics of the tumor that could determine which patients would benefit most, and controlling tumor evolution in the bone microenvironment will no doubt improve the efficacy of current therapies and significantly extend the OS of men with mCRPC.
Acknowledgements. CCL is supported by NIH R01CA143094 and U01 CA202958-01. LMC is supported by the American Cancer Society (PF-13-175-01-CSM).
Affiliations: Leah M. Cook and Conor C. Lynch are with the Department of Tumor Biology, H. Lee Moffitt Cancer Research Center and Institute, 12902 Magnolia Dr, Tampa, FL 33612, USA.
Address correspondence to:
Conor C. Lynch, H. Lee Moffitt Cancer Research Center and Institute, 12902 Magnolia Dr, Tampa, FL 33612, USA; TEL: +813-745-8094; E-mail: [email protected].
REFERENCES
- Siegel RL, Miller KD, and Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30. doi: 10.3322/caac.21332.
- Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem. 2004;91(4):718-729.
- Cook LM, Shay G, Aruajo A, Lynch CC. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev. 2014;33(2):511-525. doi: 10.1007/s10555-014-9494-4.
- Mohler JL, Armstrong AJ, Bahnson RR, et al. Prostate cancer, version 1.2016. J Natl Compr Canc Netw. 2016;14(91):19-30.
- Notelovitz M. Androgen effects on bone and muscle. Fertil Steril. 2002; 77 Suppl 4, S34-41.
- Clarke BL, Khosla S. Androgens and bone. Steroids. 2002;77(suppl 4):S34-S41. doi: 10.1016/j.steroids.2008.10.003.
- Mostaghel EA. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag Res. 22014;6:39- 51. doi: 10.2147/CMAR.S39318.
- de Bono JS, Logothetis CJ, Molina A, et al; COU-AA-302 Investigators. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995-2005. doi: 10.1056/NEJMoa1014618. doi: 10.1056/NEJMoa1014618.
- Ryan CJ, Smith MR, de Bono JS, et al; COU-AA-302 Investigators. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138-148. doi: 10.1056/NEJMoa1209096.
- So A, Chin J, Fleshner N, Saad F. Management of skeletal- related events in patients with advanced prostate cancer and bone metastases: incorporating new agents into clinical practice. Can Urol Assoc J. 2012;6(6):465-470. doi: 10.5489/cuaj.12149.
- Logothetis CJ, Basch E, Molina A, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 2012;13(12):1210-1217. doi: 10.1016/S1470-2045(12)70473-4.
- Scher HI Fizazi K, Saad F, et al; AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13);1187-1197.
- Loriot Y, Miller K, Stemberg CN, et al. Effect of enzalutamide on health-related quality of life, pain, and skeletal-related events in asymptomatic and minimally symptomatic, chemotherapy- naive patients with metastatic castration-resistant prostate cancer (PREVAIL): results from a randomised, phase 3 trial. Lancet Oncol. 2016;16(5):509-521. doi: 10.1016/S1470-2045(15)70113-0.
- Tokai Pharmaceuticals announces clinical update [press release]. Boston, MA: Tokai Pharmaceuticals; July 26, 2016. http://investors.tokaipharmaceuticals.com/phoenix.zhtml?c=252857&p=irol- newsArticle&ID=2188163. Accessed October 7, 2016.
- Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028-1038. doi: 10.1056/NEJMoa1315815.
- Maughan BL, Antonarakis ES. Clinical relevance of androgen receptor splice variants in castration-resistant prostate cancer. Curr Treat Options Oncol. 2015;16(12):57. doi: 10.1007/s11864-015-0375-z.
- Ojemuyiwa MA, Madan RA, Dahut WL. Tyrosine kinase inhibitors in the treatment of prostate cancer: taking the next step in clinical development. Expert Opin Emerg Drugs. 2014; 19(4):459- 470. doi: 10.1517/14728214.2014.969239.
- Wan X, Corn PG, Yang J, et al. Prostate cancer cell-stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases. Sci Transl Med. 2014;6(252):252ra122. doi: 10.1126/scitranslmed.3009332.
- Nakajima A, Shimizu S, Moriya H, Yamazaki M. Expression of fibroblast growth factor receptor-3 (FGFR3), signal transducer and activator of transcription-1, and cyclin-dependent kinase inhibitor p21 during endochondral ossification: differential role of FGFR3 in skeletal development and fracture repair. Endocrinology.2003;144(10):4659-4668.
- Araujo JC, Mathew P, Armstrong AJ, et al. Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1-2 study. Cancer. 2012;118(1):63-71. doi: 10.1002/cncr.26204.
- Araujo JC, Trudel GC, Saad F, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 2013;14(13):1307-1316. doi: 10.1016/S1470-2045(13)70479-0.
- Garcia-Gomez A, Ocio EM, Crusoe E, et al. Dasatinib as a bone- modifying agent: anabolic and anti-resorptive effects. PLoS One. 2012;7(4):e34914. doi: 10.1371/journal.pone.0034914.
- Smith MR, Sweeney CJ, Corn PG, et al. Cabozantinib in chemotherapy-pretreated metastatic castration-resistant prostate cancer: results of a phase II nonrandomized expansion study. J Clin Oncol. 2014;32(30): 3391-3399. doi: 10.1200/JCO.2013.54.5954.
- Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31(4):412-419. doi: 10.1200/JCO.2012.45.0494.
- Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34(25):3005-3013. doi: 10.1200/JCO.2015.65.5597.
- Schiewer MJ, Goodwin JF, Han S, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov.2012;2(12):1134-1149. doi: 10.1158/2159-8290.CD-12-0120.
- Goodwin JF, Kothari V, Drake JM, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell. 2015;28(1):97-113. doi: 10.1016/j. ccell.2015.06.004.
- Kishi Y, Fujihara H, Kawaquchi K, et al. PARP inhibitor PJ34 suppresses osteogenic differentiation in mouse mesenchymal stem cells by modulating BMP-2 signaling pathway. Int J Mol Sci. 2015;16(10):24820-24838. doi: 10.3390/ijms161024820.
- Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697-1708. doi: 10.1056/NEJMoa1506859.
- Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration- resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813-822. doi: 10.1016/S0140-6736(10)62344-6.
- Khan MA, Partin AW. Bisphosphonates in metastatic prostate cancer. Rev Urol. 2003;5(3):204-206.
- Saad F, Gleason GM, Murray R, et al; Zoledronic Acid Prostate Cancer Study Group. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94(19):1458-1468.
- Saad F, Gleason DM, Murray R, et al; Zoledronic Acid Prostate Cancer Study Group. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96(11):879-882.
- Ekelund S, Nygren P, Larsson R. Guanidino-containing drugs in cancer chemotherapy: biochemical and clinical pharmacology. Biochem Pharmacol. 2001;61(10):1183-1193.
- Parker C, Nilsson S, Heinrich D, et al; ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223. doi: 10.1056/NEJMoa1213755.
- Sonpavde G, Di Lorenzo G, Higano CS, Kantoff PW, Madan R, Shore ND. The role of sipuleucel-T in therapy for castration- resistant prostate cancer: a critical analysis of the literature. Eur Urol. 2012;61(4):639-647. doi: 10.1016/j.eururo.2011.10.027
- Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411-422. doi: 10.1056/NEJMoa1001294.
- Crawford ED, Petrylak DP, Higano CS, et al. Optimal timing of sipuleucel-T treatment in metastatic castration-resistant prostate cancer. Can J Urol. 2015;22(6):8048-8055.
- Madan RA, Arlen PM, Mohebtash M, Hodge JW, Gulley JL. Prostvac-VF: a vector-based vaccine targeting PSA in prostate cancer. Expert Opin Investig Drugs. 2009;18(7):1001-1011. doi: 10.1517/13543780902997928.
- Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099-1105. doi: 10.1200/ JCO.2009.25.0597.
- Zhang K, Kim S, Cremasco V, et al. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Res. 2011;71(14):4799-4808. doi: 10.1158/0008-5472.CAN-10-3922.
- Kwon ED, Drake CG, Scher HI, et al; CA184-043 Investigators. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700-712. doi: 10.1016/S1470-2045(14)70189-5.
- Capietto AH, Faccio R. Immune regulation of bone metastasis. Bonekey Rep. 2014;3:600. doi: 10.1038/bonekey.2014.95.
- Fellner C. Ipilimumab (yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. P T. 2012;37(9):503-530.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. [erratum appears in N Engl J Med. 2010;363(13):1290]. N Engl J Med. 2010;363(8):711- 723. doi: 10.1056/NEJMoa1003466.
- Graff JN, Puri S, Bifulco CB, Fox BA, Beer TM. Sustained complete response to CTLA-4 blockade in a patient with metastatic, castration-resistant prostate cancer. Cancer Immunol Res. 2014;2(5):399- 403. doi: 10.1158/2326-6066.CIR-13-0193.
- Modena A, Ciccarese C, Iacovelli R, et al. Immune checkpoint inhibitors and prostate cancer: a new frontier? Oncol Rev. 2016;10(1):293. doi: 10.4081/oncol.2016.293.
- Drake JM, Graham NA, Lee JK, et al. Metastatic castration- resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A. 2013;110(49):E4762-E4769. doi: 10.1073/pnas.1319948110.
- Gatenby RA. A change of strategy in the war on cancer. Nature. 2009;459(7246):508-509. doi: 10.1038/459508a.
- Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra324. doi: 10.1126/ scitranslmed.aad7842.
- Araujo A, Cook LM, Lynch CC, Basanta D. An integrated computational model of the bone microenvironment in bone- metastatic prostate cancer. Cancer Res. 2014;74(9):2391-2401. doi: 10.1158/0008-5472.CAN-13-2652.
- Cook LM, Araujo A, Pow-Sang JM, Budzevich MM, Basanta D, Lynch CC. Predictive computational modeling to define effective treatment strategies for bone metastatic prostate cancer. Sci Rep. 2016;6:29384. doi: 10.1038/srep29384.
- Gallaher J, Cook LM, Gupta S, et al. Improving treatment strategies for patients with metastatic castrate resistant prostate cancer through personalized computational modeling. Clin Exp Metastasis. 2014;31(8):991-999. doi: 10.1007/s10585-014-9674-1.


