Background
Immunotherapy is a rapidly evolving field with the goal of using the patients’ immune system to attack cancer. Much attention in the field has been given to inhibitory check- points, whereby T cells that have upregulated inhibitory molecules such as CTLA-4, PD-1/PD-L1, TIM-3 (T0cell immunoglobulin mucin-3), and LAG-3 can effectively turn down effector functions such as cytokine release and cytotoxicity—both of which are required to effectively kill tumor cells. This ratcheting down of effector function can be impeded by targeting these inhibitory molecules through the administration of monoclonal antibodies (mAbs) known as checkpoint inhibitors.
However, despite the success of checkpoint blockade with biologics such as anti-CTLA-4 (aCTLA-4) and anti-PD-1 (aPD-1) mAbs, a significant proportion of patients that receive these treatments do not develop therapeutic responses, which highlights the need for more effective therapies.
Optimal T-cell activation requires 2 events: signal 1, which is recognition of the cognate peptide/major histocompatibility complex (MHC) by a specific T-cell receptor (TCR), and signal 2, which is ligation of the CD28 costimulatory receptor by its ligands B7.1/B7.2 (CD80/CD86). Together, these are commonly known as T-cell priming. However, in addition to the initial priming event, further signaling is necessary to drive T-cell differentiation and potentiate the development of effector and memory cell subsets; these events enable robust tumor killing activity, in vivo. The tumor necrosis factor receptors (TNFRs), including glucocorticoid-induced TNFR (GITR; CD357), CD27, OX40 (CD134), and 4-1BB (CD137), are a family of proteins responsible for transducing these additional costimulatory signals. In order to harness this important signaling cascade, agonist mAbs and specific ligand complexes have been developed that can engage with these TNFRs and activate downstream events. In this review, we will discuss the biology of TNFRs, the current preclinical and clinical trials targeting these receptors, and potential synergy with other therapies in order to enhance anti-tumor immunity.
CD27
CD27 costimulation is initiated via its ligand, CD70, a homotrimeric type II membrane protein that is most commonly expressed on antigen presenting cells (APCs) after immune activation.1-6 Like other TNFR family members, CD27 is expressed on naïve CD4+ and CD8+ T cells; however, CD27 differs from other TNFR family members in that it is commonly shed from the cell surface following T-cell activation. Furthermore, this soluble form can be detected and used as a diagnostic marker.7 The cytoplasmic tail of CD27, similar to other costimulatory TNFRs, contains motifs that bind TNFR-associated factors (TRAFs), and the subsequent ubiquitination of TRAFs activates both canonical and noncanonical nuclear factor kappa B (NF-KB) pathways as well as the c-Jun-N-terminal kinase (JNK)-signaling cascade.8-10 Downstream signaling includes expression of the master transcription factor T-bet, which facilitates Th1 differentiation.11-13 These Th1 cells can subsequently promote CD8+ T-cell effector differentiation through promotion of proliferation and survival, however these cells are neither required nor sufficient to support CD8+ T-cell differentiation.13-18 The importance of the CD27 signaling pathway has been demonstrated in vivo utilizing transgenic (Tg) mouse models where either B cells or dendritic cells (DCs) constitutively express the CD27 ligand, CD70.19, 20 These mice develop increased numbers of effector CD4+ and CD8+ T cells that protect against lethal challenge of EL-4 lymphoma and B16 melanoma tumors, both of which are poorly immunogenic.20, 21 In fact, when Tg mice with CD70-expressing DCs were intravenously administered MHC class I-restricted ovalbumin peptide in the absence of adjuvants, an induction of long-lived effector CD8+ T cells was detected, whereas in wild-type (WT) mice, this treatment induced tolerance.20 Similarly, the generation of cytotoxic T cells (CTLs) in WT mice can be achieved using a soluble recombinant form of CD70 to trigger through CD27.16 These preclinical data set the groundwork for further translational work targeting the CD27:CD70L axis.
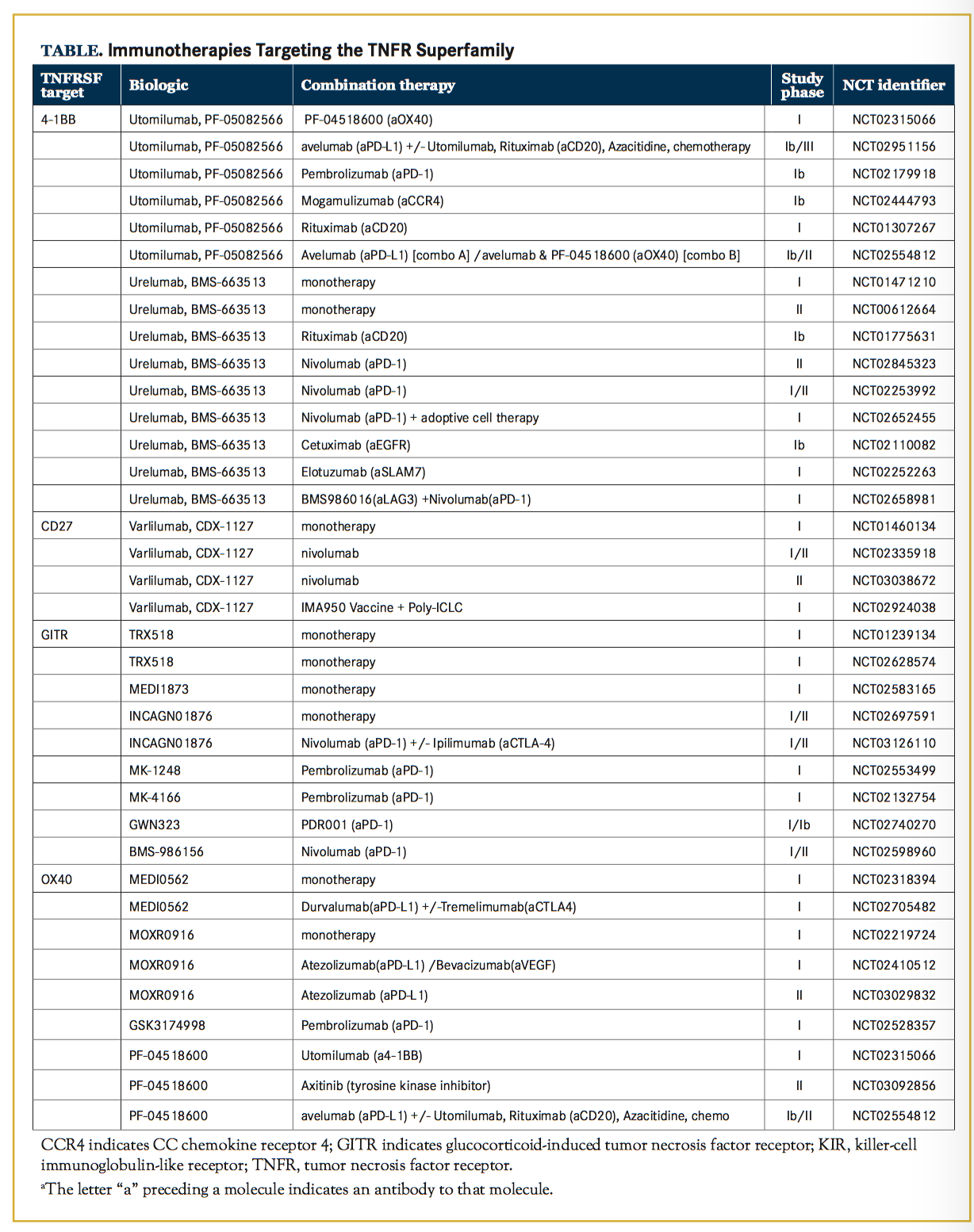
Current Clinical and Preclinical Biologics Varlilumab is a first-in-class fully human agonist IgG1 mAb that is directed against CD27 (aCD27), which has been shown to induce T-cell proliferation and production of Th1 effector cytokines such as interferon-gam- ma (IFN-gamma ) in vitro and in vivo in preclinical work.22, 23 Varlilumab has been tested in a phase I clinical trial as a monotherapy (NCT01460134) (Table) in patients with advanced B-cell lymphomas or solid tumors. In patients with B-cell lymphomas, no dose-limiting toxicities (DLTs) were reported, and furthermore, the 1 patient that experienced a complete response (CR) was subsequently found to have the highest CD27 expression level of all patients included, as determined by immunohistochemistry (IHC).24 In patients with solid tumors, 1 DLT and 1 partial response (PR) in a patient with metastatic renal cell cancer were reported. Of note, evidence of CD27 stimulation as detected by chemokine induction and T-cell stimulation was found at all dosing levels.25
In murine models, treatment with aPD-1 combined with anti-CD27 (aCD27) resulted in tumor eradication in 100% of mice due to the ability of aCD27 to stimulate CTLs in lieu of CD4+ T cell help. This response was far superior to treatment with aPD-1/aCTLA-4 dual therapy.26 Interestingly, varlilumab is being tested in combination with aPD-1 mAb (nivolumab) for safety, tolerability, and efficacy in a dose escalation and expansion phase I/II trial for patients with advanced refractory solid tumors (NCT02335918). Preliminary results from the 36 patients enrolled indicate that the combination is well-tolerated and show clinical activity in a subset of patients, including 3 patients that had an objective PR by Response Evaluation Criteria In Solid Tumors (RECIST) criteria. One of these patients experienced a 94% decrease in target lesion diameter and a progression-free survival (PFS) of 19+ months. Interestingly, compared to their baseline biopsy, posttreatment these patients had an increase in PD-L1 expressing tumors that correlated with increased CD8+ T-cell infiltration and decreased circulating regulatory T cells (Tregs).27 Another clinical trial testing the varlilumab and nivolumab combination is slated to begin enrolling patients with aggressive B-cell lymphomas December 2017 (NCT03038672). In addition to response rates and survival times, this study will measure CD27 expression and investigate changes in peripheral and intratumoral immune cells by mass cytometry, which has the capability of evaluating over 40 markers on an individual cell, and by IHC. This addition- al monitoring may shed light on how T cell costimulation synergizes with checkpoint blockade (NCT03038672).
4-1BB
4-1BB (CD137) has broad expression, which includes a vast array of cell types within the hematopoietic system, those of neuronal origin, as well as subtypes of lymphomas and leukemias.28 In particular, both CD4+ and CD8+ T cells transiently upregulate 4-1BB following activation.29 When 4-1BB ligand (4-1BBL; CD137L) engages 4-1BB, the receptor trimerizes and initiates a signaling cascade that activates downstream pathways including JNK and NF-kB.30-33 In a way similar to the action of CD27, the activation of these pathways induces increased proliferation, survival, and execution of effector functions such as cytokine release.34-39
In addition, costimulatory signals transduced through 4-1BB aide DC maturation.40 Melero et al first demonstrated that treatment with 4-1BB agonists could eradicate tumors in vivo, an effect mediated primarily by CTLs, which marked 4-1BB as an attractive immunotherapy candidate.41
Current Clinical and Preclinical Biologics
Urelumab, an engineered IgG4 mAb, and utomilumab, an IgG2 isotype mAb, are both fully human agonist anti-4-1BB mAbs (a4-1BB) currently in clinical trials.42 Urelumab’s initial translation to the clinic was stalled due to adverse events that included grade 4 liver toxicities, which resulted in termination of the trials (NCT00309023, NCT00612664).43 Further investigation identified interleukin (IL)-27 expression by myeloid cells in the liver as the process responsible for recruitment and activation of T cells, which was mediating liver damage.44 Subsequently, in a phase I safety trial including patients with either advanced solid tumors or relapsed/refractory B cell non-Hodgkin lymphoma (NCT01471210), urelumab was found to have a maximum tolerated dose of 0.1 mg/kg given every 3 weeks. Now, it is once again being investigated, this time in concert with other immunotherapies.42 For instance, urelumab is being evaluated for safety when given in combination with checkpoint inhibition (aPD-1/nivolumab; NCT02253992, NCT02845323, NCT02658981), cell-specific depleting anti- bodies (Abs) (aCD20/rituximab; NCT01775631), blocking Abs (aEGFR/cetuximab; NCT02110082), adoptive cell therapy (NCT02652455), or mAbs that modulate natural killer (NK) cell responses (aKIR/lirilumab, anti-SLAMF7/ elotuzumab; NCT02252263).
Utomilumab was first evaluated in a phase I trial in combination with rituximab. No severe adverse events were re- ported and 2 patients achieved PRs and 2 patients achieved CRs that were durable for >2 years (NCT01307267).45 Utomilumab is now under investigation in combination with other mAbs, including costimulatory molecules such as an OX40 agonist mAb (aOX40) (NCT02315066) and checkpoint inhibitors such as aPD-1 mAb (NCT02179918); and, in a triple combination, with avelumab (aPD-L1) plus aOX40 mAb (NCT02554812).
Additionally, new technology is being developed whereby checkpoint mAbs and agonist mAbs are bound to nanoparticles; this is termed an “immunoswitch.” In one trial, the immunoswitch bound with a4-1BB and aPD-L1 was superior in terms of reducing tumor burden and in- creasing survival compared to the control treatment, which was soluble a4-1BB and aPD-L1 given concurrently.46 Also in development is PRS-343, a 4-1BB agonist mAb fused to a variant of trastuzumab, which is a mAb targeting HER2, in an effort to focus immune responses toward tumor cells and away from healthy cells. PRS-343 was shown to reduce tumor growth and to elicit increased tumor infiltrating lymphocytes in a humanized mouse model. It will be interesting to see what other novel agents are capable of harnessing T-cell costimulation while also preventing the inhibitory effects of immune checkpoints.47
The advent of immunotherapy has not abrogated the need for traditional cancer therapies. Indeed, administration of radiation therapy (RT) and chemotherapeutic drugs remains the current standard of care for numerous cancer types. Preclinical models are helping to investigate whether adding a4-1BB to either RT or chemotherapy boosts treatment efficacy. For instance, highly radiosensitive gliomas were treated in murine models with RT in combination with systemic a4-1BB, which lead to increased T-cell infiltrate, reduced tumor burden, and increased survival.48 These outcomes were also observed in a murine glioma model following treatment of focal RT and CTLA-4 blockade—an effect that was enhanced by the addition of a4-1BB. This illustrated the importance of providing costimulation to bring about optimal therapeutic outcomes.49 In addition, multiple murine tumor models have demonstrated tumor regression due to treatments that combine a4-1BB with such chemotherapies as 5-fluorouracil,50 cisplatin,51 and cyclophosphamide.52
Other combination immunotherapy strategies include the use of therapeutic cancer vaccines or oncolytic viruses that either express T-cell–modulating agents themselves or are administered in conjunction with immunotherapies. An adenovirus expressing CD40L and 4-1BBL is currently being developed, and early results show its ability to induce T-cell expansion and tumor regression.53 Another strategy is DC-based vaccination, whereby DCs are pulsed with tumor antigens in order to prime T cells; which induces anti-tumor effects, including tumor regression and Th1 cytokine production (IL-2 and IFN-gamma). When DC-based vaccination was combined with a4-1BB in multiple murine models, greater anti-tumor responses were observed compared to DC-based vaccination alone.54-56 Bartkowiak et al57 reported that the addition of a4-1BB to DC-based vaccination was more efficacious in promoting tumor regression when given with an HPV+ peptide-based cancer vaccine than the addi- tion of aCTLA-4. A melanoma-specific tyrosinase related protein-2 (Trp2) peptide vaccine, when combined with TLR9 stimulation and a4-1BB, promoted increased T-cell infiltration and tumor eradication.58 However, in this climate of in- vestigating new combinatorial strategies, more is not always better. McKee et al59 recently found that while the addition of a4-1BB to an alpha-galactosylceramide–loaded irradiated tumor-cell vaccine led to increased protection against tumor rechallenge, the effects were diminished upon co-administra- tion of aPD-1, as evidenced by a reduction in effector CD8+ T cells. Although the authors do not provide a mechanism for this seeming anomaly, one could postulate that repeated T-cell activation could lead to activation-induced cell death.
In addition to the development of agonist a4-1BB mAbs, research has been focused toward developing both soluble and tumor-expressing 4-1BBL agonists. Multiple preclinical studies have shown that tumor cells engineered to express 4-1BBL elicited anti-tumor immune responses.28 Tetramers composed of 4-1BBLs bound to streptavidin (SA-4-1BBL) were tested in a murine model. SA-4-1BBL not only induced anti-tumor responses, but also induced levels of T-cell prolif- eration comparable to those produced with agonist a4-1BB mAb treatment.60 The authors also showed that the effect was not Fc-receptor or complement dependent; however, they did find a dramatic increase in non-specific T-cell activation.60 Furthermore, these experiments also revealed that SA-4-1BBL treatment created less toxicity than a4-1BB, highlighting one prominent advantage of this strategy. Moreover, the addition of 4-1BBL to therapeutic cancer vaccines lead to increased efficacy and tumor eradication.61-63 Lastly, the Gilboa laboratory has engineered a novel method to trigger 4-1BB through the use of aptamers, which are single-stranded oligonucleotides designed to bind target proteins.64 These aptamers can be enhanced by conjugation to a wide range of molecules; these could include small interfering ribonucleic acids (siRNAs) or even additional aptamers that are targeting motifs associated with tumor tissue.65-68
OX40
OX40 (CD134) is up-regulated on CD4+ and CD8+ T cells 12 to 24 hours following TCR ligation; it is downregulated 48 to 96 hours later.69-71 Similarly, OX40 ligand (OX40L) is transiently upregulated on activated APCs. Although OX40 expression is transient on both CD4+ and CD8+ T cells, CD4+ T cells maintain this expression for a longer duration then CD8+ T cells.72 Notably, TCR ligation alone is insufficient to induce optimal upregulation of OX40. Instead, IL-2/IL-2R signaling augments OX40 expression through JAK3-mediated activation of STAT3 and STAT5.73, 74 Some subsets of CD4+ T cells, like follicular helper T cells, constitutively express OX40, and their fate is initially determined by the interaction between primed CD4+ T cells and DCs, whereby the OX40:OX40L interaction leads to the upregulation of CXCR5.75 Murine Tregs constitutively express OX40; however, on human Tregs, OX40 is only induced after activation.76 Importantly, administration of OX40 agonists enhances CD4+ and CD8+ T cell expansion— presumably due to increased T cell survival as opposed to increased proliferation.77, 78 OX40 signaling also promotes T- cell differentiation; for example, OX40 ligation on primed CD8+ T cells leads to increased granzyme B expression and cytolytic activity, thus skewing toward an effector phenotype.79, 80 Soluble OX40L agonists are also being investigated as biologics. Several preclinical models have highlighted an Fc-OX40L fusion protein’s anti-tumor effects, including proliferation of tumor-infiltrating T cells, tumor regression, and increased survival.81-83
Additionally, tumor-specific T cells accumulate at the tumor site in vivo.84 The formation of antigen-specific memory T cells is dependent upon TRAF2 binding the cytoplasmic tail of OX40, which signals downstream through NF-kB.85 The NF-kB inducing kinase was revealed to be necessary for OX40 costimulation via the noncanonical NF-kB pathway.86 Anergy induction can occur due to continuous strong TCR stimulation by tumor-expressed self-antigens in the absence of costimulation. However, OX40 ligation reversed CD8+ T-cell anergy of tumor-reactive cells, which lead to their increased survival and tumor regression in murine models.87 Furthermore, OX40 engagement on intratumoral FoxP3+ Tregs decreases their immunosuppressive effects within the tumor microenvironment.70, 88-91 This reduction in Treg effectiveness may be due to prevention of naïve CD4+ T cell to Treg conversion,92 reduction in FoxP3 expression,89 and/or FC receptor (FcR)-mediated depletion of intratumoral Tregs.93, 94 However, there are reports that the cytokine milieu of the microenvironment can drive Treg expansion.90, 95 Despite these observations of varying effects on Tregs by antibody OX40 (mOX40) mAb, which are likely due to differences in tumor models and the inflammatory milieu present in these different systems, administration of agonist aOX40 mAb is extremely promising due to its ability to elicit anti-tumor responses, thus warranting further investigations in clinical trials.
Current Clinical and Preclinical Biologics
The first-in-human phase I clinical trial with an agonist aOX40 mAb was tested as a monotherapy in patients with advanced cancer (NCT01644968). Since this agent was originally developed as a mouse anti-human aOX40 mAb, patients received only 3 doses because they generated human anti-mouse antibodies against the murine IgG1 domain. However, after only one treatment cycle, an impressive 12 of 30 patients experienced regression of at least one metastatic lesion.96, 97 In addition, the treatment was generally well tol- erated, and the only grade 3/4 event reported was transient lymphopenia.97 Recapitulating preclinical results, patients treated with aOX40 mAb also had a significant increase in proliferation among effector CD4+ and CD8+ T cells, but not CD4+FoxP3+ Tregs.97 Preliminary phase I results testing the humanized IgG1 agonist aOX40 mAb, MEDI0562, in patients with advanced solid tumors revealed no DLTs and of 32 patients evaluable for response, an objective response (irRECIST and RECIST 1.1) was observed in one patient with a response duration of 16 weeks (NCT02705482).98
The agonist aOX40 mAb MEDI0562 is also being tested in combination with either aPD-L1 mAb (durvalumab) or aCTLA-4 mAb (tremelimumab) (NCT02705482). The combination of aOX40 and aCTLA-4 mAbs is akin to “stepping on the gas” with costimulatory OX40 agonists while simultaneously “removing the brakes” with check- point blockade. This rationale has been tested in pre- clinical models including a poorly immunogenic prostate cancer model and a sarcoma model. Findings from both models demonstrate that combined aOX40/aCTLA-4 mAb therapy improved tumor regression and survival.99 Interestingly, the addition of IL-4 blockade to this combi- nation therapy further enhanced the anti-tumor response, likely due to suppression of an aberrant Th2 CD4 T cell response induced following aOX40/aCTLA-4 therapy.99 Th1 polarization could also be restored when aOX40/ aCTLA-4 therapy was combined with tumor antigen-specific vaccinations, which lead to enhanced IFN-gamma production and tumor regression in a mammary carcinoma model.100
Other clinical trials combining agonist aOX40 mAbs with checkpoint blockade include the investigation of MOXRO0916, currently being tested in phase I/II clinical trials in combination with aPD-L1 mAb atezolizumab (NCT02410512, NCT03029832). No DLTs have been reported (NCT02410512), and the dose-expansion phase has been set for MOXR0916 300 mg + atezolizumab 1200 mg every 3 weeks.101 This study also includes triple dose escalation + expansion arms investigating the agonist/ checkpoint blockade in combination with bevacizumab, a recombinant humanized anti-VEGF monoclonal antibody (NCT02410512). GSK3174998 is a humanized IgG1 agonist aOX40 mAb being evaluated as a monotherapy with aPD-1 pembrolizumab (NCT02528357); initial monotherapy cohorts were completed without dose limiting toxicities.102 The fourth OX40 agonist currently being developed is PF- 04518600 (NCT02315066, NCT02554812, NCT03092856). In monotherapy cohorts, similar to other OX40 agonists currently in clinical trials, PF-04518600 was also well tolerated, and 27 of 48 patients achieved either PR (2 patients) or SD (25 patients).103 PF-04518600 is being tested in combination with a4-1BB agonists (NCT02315066) and as well as a phase 1b/II study of various combinations that all include aPD-L1 mAb (avelumab): combination A (avelumab and 4-1BB agonist, PF-05082566); combination B (avelumab and OX40 agonist, PF-04518600); combination C (avelumab and anti-cytokine colony stimulating factor 1 mAb, PD 0360324); and combination D (avelumab and 4-1BB and OX40 agonists, NCT02554812). In addition to combinations with additional immunotherapies, PF-04518600 will be tested for efficacy when combined with the tyrosine kinase inhibitor axitinib in patients with kidney cancer (NCT03092856).
Another interesting combination involves GR-MD-02, a drug that specifically inhibits galectin-3. Galectin-3 is upregulated in numerous cancers and its expression correlates with increased metastatic potential and poor patient outcomes. Additionally, galectin-3 can play a role in immune suppression by interfering with TCR accessibility and recruitment of myeloid derived suppressor cells.104 Preclinical testing revealed that the addition of GR-MD-02 to an agonist OX40 mAb or to checkpoint inhibitors including aCT- LA-4 or aPD-1 mAbs led to increased survival and tumor regression compared to immunotherapy alone.104 These data provided the rationale for further testing of GR-MD-02 plus checkpoint blockade (ipilimumab or pembrolizumab) in phase I clinical trials for patients with advanced cancer (NCT02117362, NCT02575404).
GITR
GITR (CD357) is constitutively expressed at high levels on Tregs and minimally expressed on naïve and memory T cells.105-107 However, similar to OX40, GITR is upregulated on effector T cells 24 hours post activation due to signaling cascades downstream of NF-kB.85,86,108 Additionally, there is an increase of FoxP3-mediated GITR expression on Tregs upon activation. Interestingly, activated Tregs are the immune subset with the highest GITR expression, and thus are crucial to study, to further understand the potential modes of action for GITR biologics currently under development.109 GITR is expressed on NK cells and moderately expressed on activated macrophages and DCs.110 The ligand for GITR, GITRL, is expressed on activated APCs111, 112 and on endothelial cells, which can express high levels of GITRL in the presence of type I interferon.113 GITR engagement by GITRL enhances T cell proliferation and effector function by upregulating CD25 and inducing cytokine (IL- 2/IFN-gamma) expression.114-118
Current Clinical and Preclinical Biologics
Initial preclinical studies were conducted using an agonist rat anti-mouse IgG2a aGITR mAb, (clone DTA-1).105,106 Agonist DTA-1 administration expands the number of effector CD8+ T cells with increased cytokine production; and at the same time, it abates Treg-mediated immune suppression.107 Initial hypotheses regarding this mechanism include reduced FoxP3 expression119,120 and Treg depletion due to FcgR activity.119,121,122 Recently, new evidence by Mahne et al123 shed light on this outstanding question using a Treg fate-mapping approach, wherein they found that Tregs were indeed depleted due to DTA-1 treatments. Furthermore, highly activated Tregs were preferentially targeted. A 5-part dose-escalation study is currently recruiting patients with advanced solid tumors to determine the maximum tolerated dose (MTD) of MK-4166, which binds an epitope closely approximating the epitope bound by DTA-1.124 One cohort of the study will determine the MTD for MK-4166 in combi- nation with pembrolizumab (NCT02132754). An additional GITR agonist mAb, MK-1248, is also being evaluated alone or in combination with pembrolizumab in a phase I trial (NCT02553499). The first GITR agonist (TRX518) to be evaluated in humans is a humanized aglycosyl IgG1 nondepleting mAb; it was evaluated in a phase I study in stage III/IV melanoma patients. While the dose escalation study found little toxicity (up to 8 mg/kg), the study also showed little efficacy with only 4 patients achieving the best clinical response of stable disease (n=28) (NCT01239134).125 A subsequent 2-part phase I open-label trial of TRX518 is being conducted in patients with advanced solid tumors (NCT02628574). Moreover, the role of antibody-dependent cell-mediated cytotoxicity in depleting Tregs in addition to GITR agonism will be interrogated by 2 humanized IgG1 aGITR mAbs both of which engage Fc receptors and went into phase I trials in 2016 (INCAGN01876, NCT02697591 and MEDI1873, NCT02583165).107,126 A phase I/II trial combining INCAGN01876 with nivolumab, ipilimumab, or triple combination is currently recruiting patients (NCT03126110). Another GITR agonist, BMS-986156, is being tested in a phase I trial in combination with nivolumab (NCT02598960). In addition to agonist mAbs, the use of GITRL is also being explored as a therapeutic to trigger GITR mediated costimulation. For example, MEDI1873 is a hexameric GITRL molecule that was designed to contain an IgG1 Fc domain in an effort to deplete Tregs.127 It is currently being tested in a phase I trial (NCT02583165).
Conclusions
The use of checkpoint blockade has been extensively researched in multiple cancer types and has achieved impressive results. Importantly, this reinvigoration of T cells assumes the presence of tumor-specific neoantigens capable of eliciting priming of tumor-specific T cells. Indeed, the FDA recently approved the use of pembrolizumab as a second-line treatment for all metastatic solid tumor types classified as high microsatellite instability or deficient DNA mismatch repair. Tumors with these types of mutation are associated with an increased level of neoantigens that may be recognized by tumor-reactive T cells. However, these treatments have not been successful for all patients, which highlights the critical need for additional therapeutic options. In addition to “removing the brakes” on T cells with checkpoint blockade, numerous studies are actively testing whether “stepping on the gas” by the addition of TNFR costimulatory agonists will increase tumor-specific T cell proliferation and cytolytic function, thus leading to tumor eradication (Figure). Many of the TNFR agonist mAbs/ligands currently under investigation have shown potential to stimulate T cells; however, some efficacy may be attributed to the depletion of Tregs. In addition, the sheer number of biologics coming down the pipeline warrants the rational design of clinical trials based upon the biology of the respective TNFR family members, to select optimal combinations of these new immunotherapies to bring about the best possible patient outcomes.
Author affiliations: Both Elizabeth R. Sturgill, PhD, and William L. Redmond, PhD are with the Robert W. Franz Cancer Research Center, Earle A. Chiles Research Institute, Providence Portland Medical Center, Portland, OR.
Address correspondence to: Dr William L. Redmond, Robert W. Franz Cancer Research Center, Earle A. Chiles Research Institute, Providence Portland Medical Center, 4805 NE Glisan St., 2N35, Portland, OR 97213. Tel: (503) 215-3841. E-mail: [email protected]
Financial disclosures: Elizabeth R. Sturgill reports having no competing interests. William L. Redmond has received commercial research grants, consulting fees, and/or royalties from MedImmune, Bristol-Myers Squibb, Merck, Galectin Therapeutics, Nektar Therapeutics, Aeglea Biotherapeutics, IRX Therapeutics, Tesaro, Shire, and DNAtrix.
Funding: This work was supported by the Providence Portland Medical Foundation, Susan G. Komen Grant Career Catalyst Research Grant (CCR15329664), and NIH R21CA190790.
Acknowledgements: We would like to thank Annah Rolig for her careful review of this manuscript.

References
- Bowman MR, Crimmins MA, Yetz-Aldape J, Kriz R, Kelleher K, Herrmann S. The cloning of CD70 and its identification as the ligand for CD27. J Immunol. 1994;152(4):1756-1761.
- Hintzen RQ, Lens SM, Beckmann MP, Goodwin RG, Lynch D, van Lier RA. Characterization of the human CD27 ligand, a novel member of the TNF gene family. J Immunol.1994;152(4):1762-1773.
- Tesselaar K, Gravestein LA, van Schijndel GM, Borst J, van Lier RA. Characterization of murine CD70, the ligand of the TNF receptor family member CD27. J Immunol.1997;159(10):4959-4965.
- Tesselaar K, Xiao Y, Arens R, et al. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J Immunol. 2003;170(1):33-40.
- Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol. 2007;178(3):1564-1572.
- Denoeud J, Moser M. Role of CD27/CD70 pathway of activation in immunity and tolerance. J Leukoc Biol.2011;89(2):195-203.
- Loenen WA, De Vries E, Gravestein LA, Hintzen RQ, Van Lier RA, Borst J. The CD27 membrane receptor, a lymphocyte-specific member of the nerve growth factor receptor family, gives rise to a soluble form by protein processing that does not involve receptor endocytosis. Eur J Immunol. 1992;22(2):447-455.
- Akiba H, Nakano H, Nishinaka S, et al. CD27, a member of the tumor necrosis factor receptor superfamily, activates NF-kappaB and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5, and NF-kappaB-inducing kinase. J Biol Chem. 1998; 273(21):13353-13358.
- Gravestein LA, Amsen D, Boes M, Calvo CR, Kruisbeek AM, Borst J. The TNF receptor family member CD27 signals to Jun N-terminal kinase via Traf-2. Eur J Immunol. 1998;28(7):2208-2216.
- Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation path- ways by NF-kappaB-inducing kinase. Immunity. 2004;21(4):477-489.
- Soares H, Waechter H, Glaichenhaus N, et al. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med. 2007;204(5):1095-1106.
- van Oosterwijk MF, Juwana H, Arens R, et al. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int Immunol.2007;19(6):713-718.
- Xiao Y, Peperzak V, Keller AM, Borst J. CD27 instructs CD4+ T cells to provide help for the memory CD8+ T cell response after protein immunization. J Immunol. 2008;181(2):1071-1082.
- Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nature immunology 2000;1(5):433-440.
- Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003;198(9):1369-1380.
- Rowley TF, Al-Shamkhani A. Stimulation by soluble CD70 pro- motes strong primary and secondary CD8+ cytotoxic T cell responses in vivo. J Immunol. 2004;172(10):6039-6046.
- Taraban VY, Rowley TF, Al-Shamkhani A. Cutting edge: a critical role for CD70 in CD8 T cell priming by CD40-licensed APCs. J Immunol. 2004;173(11):6542-6546.
- Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood. 2009;113(21):5167-5175.
- Arens R, Tesselaar K, Baars PA, et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFN -mediated B cell depletion. Immunity. 2001;15(5):801-812.
- Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity. 2008;29(6):934-946.
- Arens R, Schepers K, Nolte MA, et al. Tumor rejection induced by CD70-mediated quantitative and qualitative effects on effector CD8+ T cell formation. J Exp Med. 2004; 199(11):1595-1605.
- He LZ, Prostak N, Thomas LJ, et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol. 2013,191(8):4174-4183.
- Vitale LA, He LZ, Thomas LJ, et al: Development of a human monoclonal antibody for potential therapy of CD27-expressing lymphoma and leukemia. Clin Cancer Res. 2012;18(14):3812-3821.
- Ansell SM, Northfelt DW, Flinn I, et al. Phase I evaluation of an agonist anti-CD27 human antibody (CDX-1127) in patients with advanced hematologic malignancies. J Clin Oncology. 2014;32:3024-3024.
- Burris HA, Infante JR, Ansell SM, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol. 2017;35(18):2028-2036.doi: 10.1200/JCO.2016.70.1508.
- Ahrends T, Bąbała N, Xiao Y, Yagita H, van Eenennaam H, Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T-cell help in therapeutic anticancer vaccination. Cancer Res. 2016;76(10):2921-2931.
- Sanborn RE, Pishvaian MJ, Kluger HM, et al. Clinical results with combination of anti-CD27 agonist antibody, varlilumab, with an- ti-PD1 antibody nivolumab in advanced cancer patients. J Clin Oncol. 2017;35(15_suppl) abstr 3007.
- Bartkowiak T, Curran MA. 4-1BB agonists: multi-potent potentiators of tumor immunity. Front Oncol. 2015;5:117. doi: 10.3389/fonc.2015.00117. eCollection 2015.
- Dawicki W, Watts TH. Expression and function of 4-1BB during CD4 versus CD8 T cell responses in vivo. Eur J Immunol. 2004;34(3):743- 751.
- Jang IK, Lee ZH, Kim YJ, Kim SH, Kwon BS. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-kappa B. Biochem Biophys Res Commun. 1998;242(3):613-620.
- Kim HH, Kwack K, Lee ZH. Activation of c-jun N-terminal kinase by 4-1BB (CD137), a T cell co-stimulatory molecule. Mol Cells. 2000;10(3):247-252.
- Kim JO, Kim HW, Baek KM, Kang CY. NF-kappaB and AP-1 regulate activation-dependent CD137 (4-1BB) expression in T cells. FEBS Lett. 2003;541(1-3):163-170.
- Oussa NA, Soumounou Y, Sabbagh L. TRAF1 phosphorylation on Serine 139 modulates NF-kappaB activity downstream of 4-1BB in T cells. Biochem Biophys Res Commun. 2013;432(1):129-134. doi: 10.1016/j.bbrc.2013.01.073.
- Starck L, Scholz C, Dorken B, Daniel PT. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xL and c-FLIP(short) via phosphatidylinositol 3-kinase and AKT/protein kinase B. Eur J Immunol. 2005;35(4):1257-1266.
- Shuford WW, Klussman K, Tritchler DD, et al. 4-1BB costimula- tory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186(1):47-55.
- Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J Immunol. 2002;169(9):4882-4888.
- Lee HW, Nam KO, Park SJ, Kwon BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol.2003;33(8):2133-2141.
- Lee HW, Nam KO, Seo SK, Kim YH, Kang H, Kwon BS. 4-1BB cross-linking enhances the survival and cell cycle progression of CD4 T lymphocytes. Cell Immunol. 2003;223(2):143-150.
- Nam KO, Kang H, Shin SM, et al. Cross-linking of 4-1BB activates TCR-signaling pathways in CD8+ T lymphocytes. J Immunol. 2005;174(4):1898-1905.
- Kuang Y, Weng X, Liu X, Zhu H, Chen Z, Chen H. Effects of 4-1BB signaling on the biological function of murine dendritic cells. Oncol Lett. 2012;3(2):477-481.
- Melero I, Shuford WW, Newby SA, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682-685.
- Segal NH, Logan TF, Hodi FS, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal anti- body. Clin Cancer Res. 2017;23(8):1929-1936.
- Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010;37(5):508-516. doi: 10.1053/j.seminoncol.2010.09.008.
- Bartkowiak T, Jaiswal AR, Ai M, Budhani P, Ager C, Curran MA. Mechanisms underlying 4-1BB agonist antibody mediated hepatotoxicity. J Immunol. 2016;196:(suppl 1); abstr 188.
- Gopal AK, Bartlett NL, Levy R, et al. A phase I study of PF- 05082566 (anti-4-1BB)+ rituximab in patients with CD20+ NHL. J Clin Oncol. 2015;33(suppl 15);3004.
- Kosmides AK, Sidhom JW, Fraser A, Bessell CA, Schneck JP. Dual targeting nanoparticle stimulates the immune system to inhibit tumor growth. ACS Nano. 2017;11(6):5417-5429. doi: 10.1021/acsna- no.6b08152.
- Hinner MJ, Bel Aiba R-S, Schlosser C, et al: Abstract B016: Costimulatory T-cell engagement by PRS-343, a CD137 (4-1BB)/HER2 bispecific, leads to tumor growth inhibition and TIL expansion in humanized mouse model. Cancer Immunol Res. 2016; 4:B016-B016.
- Newcomb EW, Lukyanov Y, Kawashima N, et al. Radiotherapy enhances antitumor effect of anti-CD137 therapy in a mouse Glioma model. Radiat Res. 2010;173(4):426-432.
- Belcaid Z, Phallen JA, Zeng J, See AP, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long- term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One 2014, 9(7):e101764. doi: 10.1371/jour- nal.pone.0101764.
- Ju SA, Cheon SH, Park SM, et al. Eradication of established renal cell carcinoma by a combination of 5-fluorouracil and anti-4-1BB monoclonal antibody in mice. Int J Cancer. 2008;122(12):2784-2790. doi: 10.1002/ijc.23457.
- Kim YH, Choi BK, Kim KH, Kang SW, Kwon BS. Combination therapy with cisplatin and anti-4-1BB: synergistic anticancer effects and amelioration of cisplatin-induced nephrotoxicity. Cancer Res. 2008;68(18):7264-7269. doi: 10.1158/0008-5472.CAN-08-1365.
- Kim YH, Choi BK, Oh HS, Kang WJ, Mittler RS, Kwon BS: Mechanisms involved in synergistic anticancer effects of anti-4-1BB and cyclophosphamide therapy. Mol Cancer Ther. 2009;8(2):469-478. doi: 10.1158/1535-7163.MCT-08-0993.
- Eriksson E, Milenova I, Wenthe J, et al. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res. 2017;23(19):5846-5857. doi: 10.1158/1078-0432.CCR-17-0285.
- Ito F, Li Q, Shreiner AB, Okuyama R, et al. Anti-CD137 mono- clonal antibody administration augments the antitumor efficacy of dendritic cell-based vaccines. Cancer Res. 2004;64(22):8411-8419.
- Cuadros C, Dominguez AL, Lollini P-L, et al. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4-1BB monoclonal antibodies induces T cell–mediated protective immunity in Her-2/neu transgenic mice. Int J Cancer. 2005;116(6):934-943.
- Lee H, Park HJ, Sohn HJ, Kim JM, Kim SJ. Combinatorial therapy for liver metastatic colon cancer: dendritic cell vaccine and low-dose agonistic anti-4-1BB antibody co-stimulatory signal. J Surg Res. 2011;169(1):e43-e50. doi: 10.1016/j.jss.2011.03.067.
- Bartkowiak T, Singh S, Yang G, et al. Unique potential of 4-1BB agonist antibody to promote durable regression of HPV+ tumors when combined with an E6/E7 peptide vaccine. Proc Natl Acad Sci USA. 2015;112(38):E5290-E5299. doi: 10.1073/pnas.1514418112.
- Sin JI, Kim H, Ahn E, et al. Combined stimulation of TLR9 and 4.1 BB augments Trp2 peptide vaccine-mediated melanoma rejection by increasing Ag-specific CTL activity and infiltration into tumor sites. Cancer Lett. 2013;330(2):190-199. doi: 10.1016/j.canlet.2012.11.045.
- McKee SJ, Doff BL, Soon MSF, Mattarollo SR. Therapeutic efficacy of 4-1BB costimulation is abrogated by PD-1 blockade in a model of spontaneous B-cell lymphoma. Cancer Immunol Res. 2017;5(3):191-197. doi: 10.1158/2326-6066.CIR-16-0249.
- Schabowsky RH, Elpek KG, Madireddi S, et al. A novel form of 4-1BBL has better immunomodulatory activity than an agonistic anti-4-1BB Ab without Ab-associated severe toxicity. Vaccine. 2009;28(2):512- 522. doi: 10.1016/j.vaccine.2009.09.127.
- Sharma RK, Elpek KG, Yolcu ES, et al. Costimulation as a plat- form for the development of vaccines: a peptide-based vaccine containing a novel form of 4-1BB ligand eradicates established tumors. Cancer Res. 2009; 69(10):4319-4326. doi: 10.1158/0008-5472.CAN-08-3141.
- Sharma RK, Schabowsky RH, Srivastava AK, et al. 4-1BB ligand as an effective multifunctional immunomodulator and antigen delivery vehicle for the development of therapeutic cancer vaccines. Cancer Res. 2010;70(10):3945-3954. doi: 10.1158/0008-5472.CAN-09-4480.
- Sharma RK, Srivastava AK, Yolcu ES, et al. SA-4-1BBL as the immunomodulatory component of a HPV-16 E7 protein based vaccine shows robust therapeutic efficacy in a mouse cervical cancer model. Vaccine. 2010;28(36):5794-5802. doi: 10.1016/j.vaccine.2010.06.073.
- Gilboa E, McNamara J, 2nd, Pastor F. Use of oligonucleotide aptamer ligands to modulate the function of immune receptors. Clin Cancer Res. 2013;19(5):1054-1062. doi: 10.1158/1078-0432.CCR-12-2067.
- Schrand B, Berezhnoy A, Brenneman R, et al. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol Res. 2014;2(9):867-877. doi: 10.1158/2326-6066.CIR-14-0007.
- Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest. 2014;124(1):188-197.
- Sakib Hossain DM, Duttagupta P, Kortylewski M. The aptamer-siRNA conjugates: reprogramming T cells for cancer therapy. Ther Deliv. 2015;6(1):1-4. doi: 10.4155/tde.14.92.
- Pastor F, Kolonias D, McNamara JO, 2nd, Gilboa E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol Ther. 2011;19(10):1878-1886. doi: 10.1038/mt.2011.145.
- So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci USA. 2006;103(10):3740-3745.
- Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005,105(7):2845-2851.
- Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29(3):187-201.
- Cannons JL, Lau P, Ghumman B, et al. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol. 2001;167(3):1313-1324.
- Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-me- diated regulation of OX40 expression. PLoS One. 2012;7(4):e34467. doi: 10.1371/journal.pone.0034467.
- McNamara MJ, Kasiewicz, MJ, Linch SN, Dubay C, Redmond WL. Common gamma chain ( c) cytokines differentially potentiate TNFR family signaling in antigen-activated CD8 T cells. J Immunother Cancer. 2014;2:28. doi: 10.1186/s40425-014-0028-y.
- Deenick EK, Ma CS. The regulation and role of T follicular helper cells in immunity. Immunology. 2011;134(4):361-367. doi: 10.1111/j.1365- 2567.2011.03487.x.
- Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol. 2010;28:57-78. doi: 10.1146/an- nurev-immunol-030409-101243.
- Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15(3):445-455.
- Weatherill AR, Maxwell JR, Takahashi C, Weinberg AD, Vella AT. OX40 ligation enhances cell cycle turnover of Ag-activated CD4 T cells in vivo. Cell Immunol. 2001;209(1):63-75.
- Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol 2007, 179(11):7244-7253.
- Jenkins SJ, Perona-Wright G, Worsley AG, Ishii N, MacDonald A. Dendritic cell expression of OX40 ligand acts as a costimulatory, not polarizing, signal for optimal Th2 priming and memory induction in vivo. J Immunol. 2007;179(6):3515-3523.
- Sadun RE, Hsu WE, Zhang N, et al. Fc-mOX40L fusion protein produces complete remission and enhanced survival in 2 murine tumor models. J Immunother. 2008;31(3):235-245. doi: 10.1097/CJI.0b013e- 31816a88e0.
- Murphy KA, Lechner MG, Popescu FE, et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin Cancer Res. 2012;18(17):4657-4668. doi: 10.1158/1078-0432.CCR-12-0990.
- Morris NP, Peters C, Montler R, et al. Development and characterization of recombinant human Fc:OX40L fusion protein linked via a coiled-coil trimerization domain. Mol Immunol. 2007;44(12):3112-3121.
- Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68(13):5206-5215. doi: 10.1158/0008-5472.CAN-07-6484.
- Prell RA, Evans DE, Thalhofer C, Shi T, Funatake C, Weinberg AD. OX40-mediated memory T cell generation is TNF receptor-associated factor 2 dependent. J Immunol. 2003;171(11):5997-6005.
- Murray SE, Polesso F, Rowe AM, et al. NF-kappaB-inducing kinase plays an essential T cell-intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J Clin Invest. 2011;121(12):4775-4786. doi: 10.1172/JCI44943.
- Redmond WL, Gough MJ, Weinberg AD: Ligation of the OX40 co-stimulatory receptor reverses self-Ag and tumor-induced CD8 T-cell anergy in vivo. Eur J Immunol. 2009;39(8):2184-2194. doi: 10.1002/eji.200939348.
- Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205(4):825-839. doi: 10.1084/jem.20071341.
- Vu MD, Xiao X, Gao W, Degauque N, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110(7):2501-2510.
- Ruby CE, Yates MA, Hirschhorn-Cymerman D, et al. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J Immunol. 2009;183(8):4853-4857. doi: 10.4049/jimmunol.0901112.
- Keller AM, Borst J. Control of peripheral T cell survival: a delicate division of labor between cytokines and costimulatory molecules. Hum Immunol.2006;67(6):469-477.
- So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and anti- gen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179(3):1427-1430.
- Marabelle A, Kohrt H, Sagiv-Barfi I, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123(6):2447-2463.
- Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92(6):475- 480. doi: 10.1038/icb.2014.26.
- Baeyens A, Saadoun D, Billiard F, et al. Effector T cells boost regulatory T cell expansion by IL-2, TNF, OX40, and plasmacytoid dendritic cells depending on the immune context. J Immunol. 2015;194(3):999- 1010. doi: 10.4049/jimmunol.1400504.
- Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34. doi: 10.3389/fonc.2015.00034.
- Curti BD, Kovacsovics-Bankowski M, Morris N, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73(24):7189-7198.
- Glisson BS, Leidner R, Ferris RL, et al. Phase 1 study of MEDI0562, a humanized OX40 agonist monoclonal antibody (mAb), in adult patients (pts) with advanced solid tumors. Presented at: ESMO 2016 Congress. Ann Oncology. 2016, 27: abstr 1052PD.
- Redmond WL, Linch SN, Kasiewicz MJ: Combined targeting of co-stimulatory (OX40) and co-inhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust anti-tumor immunity. Cancer Immunol Res. 2014;2(2):142-53. doi: 10.1158/2326-6066.CIR-13-0031-T.
- Linch SN, Kasiewicz MJ, McNamara MJ, Hilgart-Martiszus IF, Farhad M, Redmond WL. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and pro- motes survival in tumor-bearing mice. Proc Natl Acad Sci U S A 2016, 113(3):E319-327.
- Infante JR, Hansen AR, Pishvaian MJ, et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J Clin Oncol. 2016;34(suppl; abstr 101).
- Infante J, Ahlers CM, Hodi FS, et al. Abstract CT027: A phase I, open-label study of GSK3174998 administered alone and in combination with pembrolizumab in patients (pts) with selected advanced solid tumors (ENGAGE-1). Cancer Res. 2016;76(14):abstr CT027-CT027.
- El-Khoueiry AB, Hamid O, Thompson JA et al. The relationship of pharmacodynamics (PD) and pharmacokinetics (PK) to clinical outcomes in a phase I study of OX40 agonistic monoclonal antibody (mAb) PF-04518600 (PF-8600). J Clin Oncol. 2016;35(suppl; abst 3027).
- Linch S, Kasiewicz MJ, McNamara M, Hilgart I, Farhad M, Redmond W. Galectin-3 inhibition using novel inhibitor GR-MD-02 improves survival and immune function while reducing tumor vasculature. J Immunother Cancer. 2015;3(suppl 2): P306.
- McHugh RS, Whitters MJ, Piccirillo CA, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002; 16(2):311- 323.
- Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3(2):135-142.
- Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer. 2016;67:1-10. doi: 10.1016/j.ejca.2016.06.028.
- Zhan Y, Gerondakis S, Coghill E, et al. Glucocorticoid-induced TNF receptor expression by T cells is reciprocally regulated by NF-kappaB and NFAT. J Immunol. 2008;181(8):5405-5413.
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003; 4(3):337-342.
- Clouthier DL, Watts TH. Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev. 2014;25(2):91-106. doi: 10.1016/j.cytogfr.2013.12.003.
- Hanabuchi S, Watanabe N, Wang YH, et al. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL). Blood. 2006;107(9):3617-3623.
- Krausz LT, Bianchini R, Ronchetti S, Fettucciari K, Nocentini G, Riccardi C. GITR-GITRL system, a novel player in shock and inflammation. ScientificWorldJournal.2007;7:533-566.
- Nardelli B, Zaritskaya L, McAuliffe W, et al. Osteostat/tumor necrosis factor superfamily 18 inhibits osteoclastogenesis and is selectively expressed by vascular endothelial cells. Endocrinology. 2006;147(1):70-78.
- Tone M, Tone Y, Adams E, et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci USA. 2003;100(25):15059-15064.
- Kanamaru F, Youngnak P, Hashiguchi M, et al. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol. 2004;172(12):7306-7314.
- Ronchetti S, Zollo O, Bruscoli S, et al. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol. 2004;34(3):613-622. doi: 10.1002/eji.200324804.
- Stephens GL, McHugh RS, Whitters MJ, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173(8):5008-5020.
- Ronchetti S, Nocentini G, Bianchini R, Krausz LT, Migliorati G, Riccardi C. Glucocorticoid-induced TNFR-related protein lowers the threshold of CD28 costimulation in CD8+ T cells. J Immunol. 2007; 179(9):5916-5926.
- Cohen AD, Schaer DA, Liu C, et al. Agonist anti-GITR mono- clonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One 2010;5(5):e10436. doi: 10.1371/journal.pone.0010436.
- Schaer DA, Budhu S, Liu C,et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res. 2013;1(5):320-331. doi: 10.1158/2326-6066.CIR-13-0086.
- Coe D, Begom S, Addey C, White M, Dyson J, Chai JG. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol Immunother. 2010;59(9):1367-1377. doi: 10.1007/s00262-010-0866-5.
- Bulliard Y, Jolicoeur R, Windman M, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med .2013;210(9):1685-1693. doi: 10.1084/jem.20130573.
- Mahne AE, Mauze S, Joyce-Shaikh B, et al. Dual roles for regulatory T-cell depletion and costimulatory signaling in agonistic GITR targeting for tumor immunotherapy. Cancer Res. 2017;77(5):1108-1118. doi: 10.1158/0008-5472.CAN-16-0797.
- Sukumar S, Wison D, Yu Y, et al. Characterization of MK-4166, a clinical agonistic mAb that targets human GITR and inhibits the generation and activity of Tregs. Euro J Cancer. 2016;69(suppl 1):S104.
- Koon HB, Shepard DR, Merghoub T, Schaer DA, Sirard CA, Wolchok JD: First-in-human phase 1 single-dose study of TRX-518, an anti-human glucocorticoid-induced tumor necrosis factor receptor (GITR) monoclonal antibody in adults with advanced solid tumors. J Clin Oncol. 2016;34(suppl 15;abstr 3017).
- Gonzalez AM, Breous E, Manrique ML, et al. A novel agonist antibody (INCAGN01876) that targets the costimulatory receptor GITR. Presented at: the American Association for Cancer Research Annual Meeting ; April 16-20, 2016; New Orleans, LA. Abstr 3220. agenusbio. com/wp-content/uploads/2016/08/Gonzalez_3220_AACR2016_ GITR_FINAL.pdf. Accessed October 24, 2017.
- Stewart RA, Tigue N, Ireland S, et al. MEDI1873: A novel hexameric GITRL fusion protein with potent agonsitic and immuno- modulatory activities in preclinical systems. AACR. 2016;76(suppl 14): abstr 561. http://cancerres.aacrjournals.org/content/76/14_Supplement/561. Accessed October 24, 2017.





