Introduction
Acute myeloid leukemia (AML) remains among the few hematological malignancies with no major drugs approved to market in the United States in the last decade. In fact, the only drug approved in the United States for AML in the past 4 decades was gemtuzumab ozogamicin (GO; Mylotarg); it was approved in 2001 and later voluntarily withdrawn from the market due to a concern for increased toxicity in a phase III study.1 Cytarabine and anthracycline (idarubicin or daunorubicin) regimens remain the standard frontline approach in most patients with AML.2 However, the last decade has seen major advances in the understanding of molecular leukemogenesis, of immune pathways, and of conjugated and bispecific antibody technology. These advances have provided crucial insights into disease pathophysiology and platforms leading to the development of novel therapies for AML.
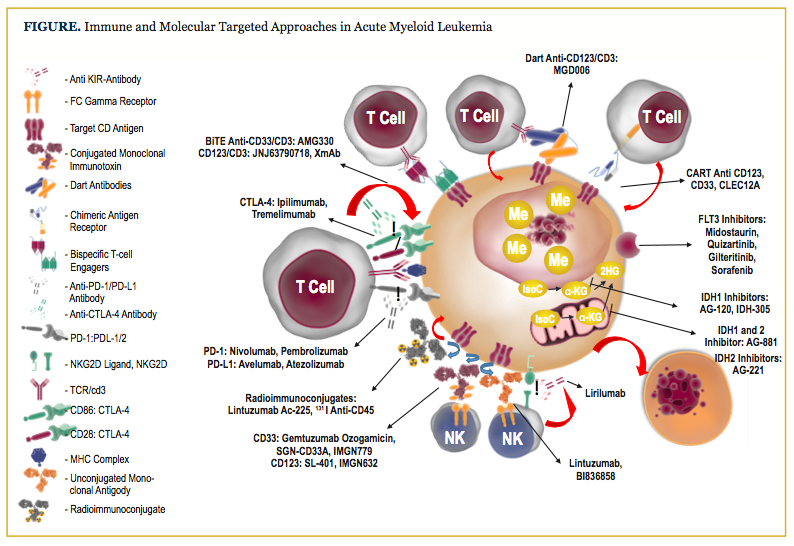
The advent of high-throughput sequencing methods has enabled characterization of recurring, prognostically informative mutations that may serve as suitable targets for small-molecule and metabolic therapies. This is exemplified by the successful targeting by novel small-molecule inhibitors of recurrent mutations and mutation-associated pathways that play a role in leukemogenesis (eg, FMS-like tyrosine kinase-3 [FLT3] and isocitrate dehydrogenase [IDH] 1/2).3 Additional small-molecule inhibitors targeting overexpressed or aberrantly regulated pathways in AML are also showing encouraging results as single agents or in combinatorial approaches (eg, inhibition of B-cell lymphoma 2 protein [BCL2], of mouse double minute 2 homolog [MDM2], and of chromosomal maintenance 1 [CRM1]) (see Figure).
Identification and targeting of leukemia-specific antigens com- pose a second major area of active research in AML. Different approaches to target these antigens, with the intent of inducing preferential cytotoxicity to leukemia blasts and potentially to leukemia stem cells, are being explored. They include monoclonal antibodies; naked or antibody drug conjugates; radioimmunoconjugates; dual-affinity retargeting antibodies; and T-cell adoptive therapy, such as chimeric antigen receptor-T (CAR-T) cells.
A third approach focuses on unleashing the patient’s own immune system to fight against leukemic cells using immune checkpoint antibodies; bispecific T-cell engager (BiTE) antibodies; and adoptively transferred natural killer (NK) cells. Clinical trial experiences with these above-mentioned therapies (Table) suggest marginal therapeutic benefit when used as single agents, but suggest additive benefit, and in many instances, synergistic benefit, when implemented in rational combinations.
Molecular Targeted Therapies in Acute Myeloid Leukemia
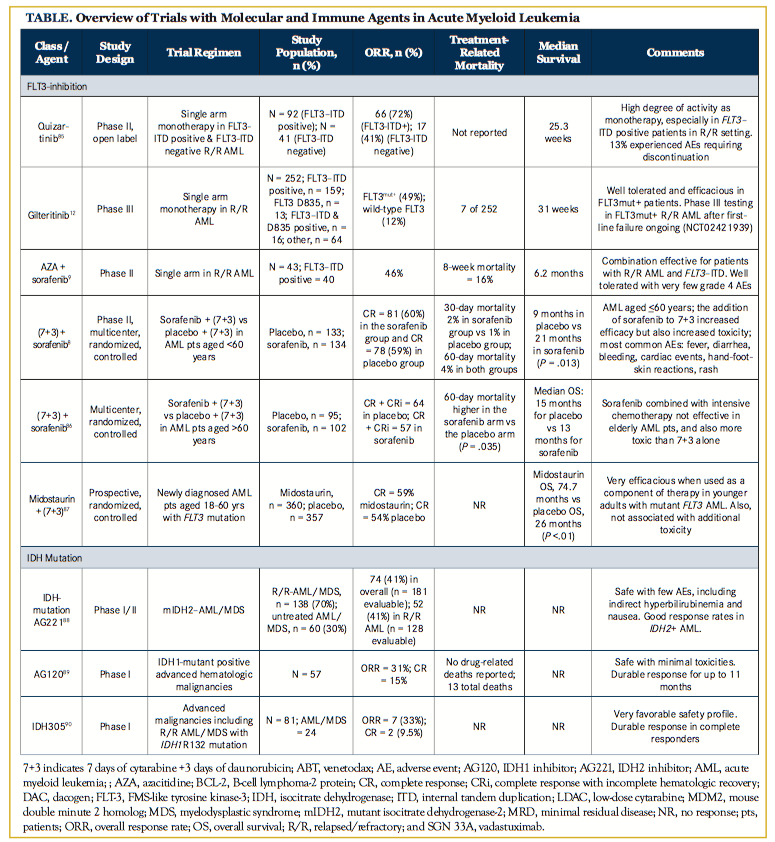
Genome-wide sequencing on large numbers of AML patients has identified recurrent mutations in genes encoding epigenetic regulators, signaling receptors, and splicing factors, as well as other genes that regulate key cellular processes.3 Notably, there are few obvious associations between a particular baseline mutational status and particular drug sensitivity, as exemplified by the sensitivity of mixed lineage leukemia (MLL)-rearranged leukemias to bromodomain inhibitors and to CDK6 inhibitors; NPM1-mutated leukemias to arsenic trioxide; and IDH2-mutated leukemias to BCL-2 inhibitors in preclinical and clinical testing.4-6 While drugs targeting AML molecular mutations—including EZH, MLL, DNMT3A, ASXL1, and TET2—are currently in preclinical or early clinical development, FLT3 and IDH inhibitors are already in an advanced phase of clinical development (Table).7
FLT3
Phase II studies have demonstrated that sorafenib, an FLT3 inhibitor, may improve response rates and event-free survival when used in combination with hypomethylating agents or cytotoxic chemotherapy.8,9 In addition, a number of other FLT3 inhibitors (quizartinib, gilteritinib, midostaurin) are currently being evaluated in phase III trials. In a large retrospective analysis, quizartinib appeared to be superior to standard-of-care regimens, both in response rates (43% vs 11%, P = .002) and median survival (in relapsed patients: 128 vs 53 days) when used in FLT3-internal tandem duplication–positive (ITD+) patients with AML who relapsed after salvage chemotherapy (SCT).10 An interim analysis of 52 patients, showed the combination of quizartinib with low-dose cytarabine (LDAC) or azacitidine to be effective, with overall response rate (ORR) of 73% and a median survival of 14.8 months.11 Gilteritinib (ASP2215) was evaluated in a phase I/II trial involving relapsed/refractory (R/R) AML (FLT3mut+ = 169) and was effective with an ORR of 52%, median duration of response of 20 weeks, and median survival of 7.8 months.12 Most recently, midostaurin received breakthrough therapy designation from the FDA for newly diagnosed FLT3-mutated AML, after demonstrating significantly improved overall survival (OS) in a randomized phase III study of induction and consolidation chemotherapy combined with midostaurin or placebo.13 Crenolanib is another orally bioavailable pan-FLT3 inhibitor with added activity against D835H and D835Y mutants. Clinical activity has been seen even in patients who have failed prior FLT3 tyrosine kinase inhibitor (TKI) therapy. A phase II single-center study evaluating crenolanib in FLT3-mutated AML suggested modest activity with complete response (CR) rates of 23% in FLT3-TKI–naïve patients and of 5% in patients who had failed prior anti-FLT3 therapy.14 The combination of crenolanib with standard cytarabine and anthracycline induction in newly diagnosed FLT3-mutant AML provided high CR rates of 88% while being well tolerated with a low incidence of AEs.15 This agent is currently undergoing evaluation in combination with other standard therapies in the treatment-naïve and salvage setting in FLT3-mutated AML (NCT02298166, NCT02626338, NCT02400281, and NCT02283177). Lestaurtinib was among the first FLT3 inhibitors to be extensively evaluated as a monotherapy and in combination with SCT. Clinical activity as a single-agent therapy was shown to be modest.16 Furthermore, its addition to SCT did not improve response rates, while it was associated with a higher frequency of severe AEs and deaths (NCT00079482).17 This agent is currently not undergoing active evaluation in FLT3-mutated AML.
IDH 1 and 2
Two first-generation IDH inhibitors—AG-221 (IDH2 inhibitor) and AG-120 (IDH1 inhibitor)—and a second-generation pan-IDH inhibitor, AG-881, are currently undergoing active study in ongoing clinical trials. Another agent, IDH-305, targeting IDH1 R132 mutation, is being evaluated in a phase I trial involving R/R AML (n = 24) and other malignancies. Preliminary data with the IDH1 and IDH2 inhibitors are encouraging, with an ORR in the 35% to 38% range and an acceptable safety profile.18 The drugs are oral, have been very well tolerated overall, and are now being evaluated in combination with 7 + 3 in younger patients with AML and with azacitidine in older patients with AML harboring IDH1 or IDH2 mutations, respectively. Differentiation is frequently seen with IDH inhibitors and often presents as increased white blood cell count, increased blasts, pulmonary infiltrates, and dyspnea. The occurrence of differentiation syndrome with IDH inhibitors does not correlate closely with the degree of leukocytosis, unlike the differentiation syndrome seen with all-trans retionic acid (ATRA) or with arsenic trioxide in acute promyelocytic leukemia (APL). The differentiation syndrome responds rapidly to steroids, and a majority of patients are able to continue therapy with the IDH inhibitors.
IDH inhibitors are differentiation-inducers and do not frequently eliminate the malignant clone, as is the case with AG-221. In contrast, most recent data suggest that AG-120 is able to affect IDH1 mutational clearance. In 1 study, among 63 patients with R/R AML (of 78 with hematological malignancy), 21 (33%) had objective responses: (CR = 10; complete response with incomplete hematologic recovery (CRi)/complete response with incomplete platelet recovery (CRp) = 8; marrow CR = 2; partial response (PR) = 1. Importantly, mIDH1 clearance by next-generation sequencing was observed in 36% of CRs and 4% of non-CRs.19 Patients with an mIDH1 clearance had improved clinical benefit from IDH inhibitors as compared with those who achieved a clinical response per International Working Group criteria but did not achieve molecular remission. Nevertheless, clonal persistence, and its therapeutic and prognostic implications for the need for continued IDH inhibitor therapy, remain unknown.
BCL2
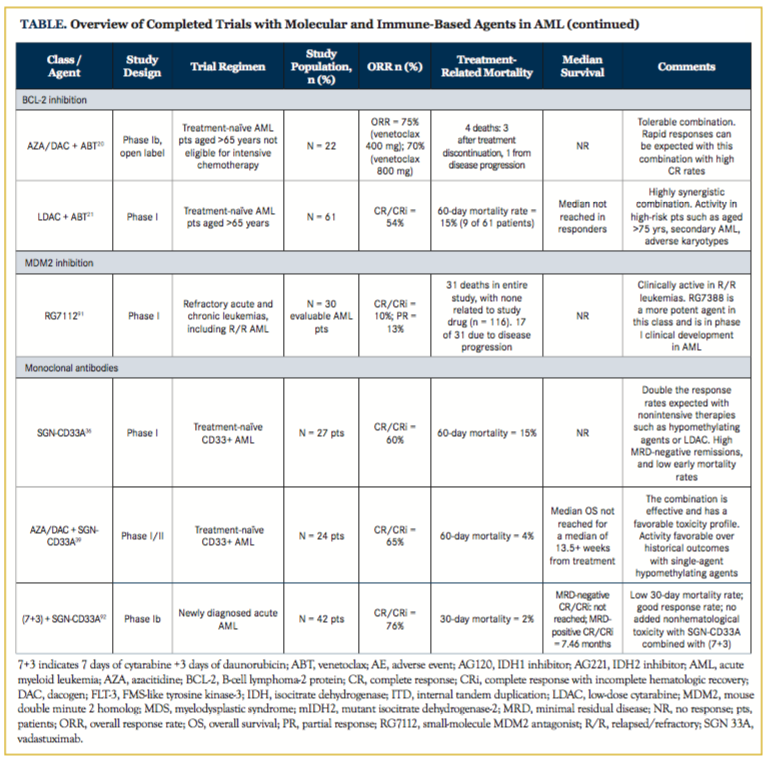
Among the BCL-2 inhibitors, venetoclax is particularly effective and is being tested in combination with other agents, including hypomethylating agents and LDAC. Venetoclax in combination with azacitidine/decitabine produced an ORR (CR/CRi/PR) of 75% in frontline older patients with AML.20 The expected response rate with azacitidine or decitabine alone is 18% to 25%, based on published data, suggesting striking synergism when the agents are combined. A phase III, randomized, registrational trial of azacitidine plus venetoclax versus azacitidine alone has begun enrollment. In a separate phase Ib study, Wei and colleagues reported on the safety and efficacy of venetoclax plus LDAC in 61 treatment-naïve patients >65 years with AML. The combination was well tolerated and produced high response rates (CR/CRi of 54%), with median survival not reached among the responders (CR/CRi/PR). Historic response rates with LDAC alone in a similar patient population have been 5% to 10%, highlighting the significant improvement when venetoclax is added to LDAC. The regimen was well tolerated. A future phase III randomized trial of LDAC with venetoclax is planned.21
Monoclonal Antibodies in Acute Myeloid Leukemia
CD33
It has been realized that leukemic stem cells exhibit phenotypic characteristics distinct from those of normal hematopoietic stem cells.22 Of the various differentially expressed cluster differentia- tion (CD) antigens on leukemic blasts, CD123, CD33, and CD56 have thus far been exploited clinically as targets for monoclonal antibody-based therapies in AML.23-26 These monoclonal antibodies may exert their anti-AML tumor effect by varied mechanisms, including antibody-mediated neutralization, delivery of toxic payload in the case of conjugated antibodies, antibody-dependent cellular cytotoxicity, complement-mediated cytotoxicity, antibody-dependent cell-mediated phagocytosis, and enhanced T-cell antitumor efficacy by increasing T cell and tumor interaction with bispecific T-cell engaging antibodies.
GO, a humanized anti-CD33 monoclonal antibody conjugated to a DNA-damaging toxin (calicheamicin), is among the most extensively studied monoclonal antibodies in AML. It was first approved in May 2000 based on a phase II trial showing a 30% response rate in patients with AML in first relapse. However, GO was subsequently voluntarily withdrawn from the market 10 years later,27-29 based on a Southwest Oncology Group (SWOG) phase III trial that demonstrated a lack of benefit and potentially increased mortality in patients who received GO with standard chemotherapy.1 However, multiple criticisms have since been raised against the design and dosing schema of this SWOG study. Since the SWOG study, 4 randomized trials conducted in Europe have demonstrated efficacy of GO in patients with AML with good- and intermediate-risk cytogenetics.30-34 Additionally, the higher incidence of veno-occlusive disease with the single high dose of 9 mg/m2 was significantly reduced by administering multiple split doses of 3 to 6 mg/m2 in these trials. Based on these data, the use of GO is being reassessed by the European and US drug agencies and is simultaneously being further evaluated in de novo, relapsed, and pretransplant settings in clinical trial (NCT01869803, NCT02473146, NCT02221310). SGN-CD33A (vadastuximab), an anti-CD33 antibody conjugated to pyrrolobenzodiazepine, is a newer CD33-targeted monoclonal that demonstrated cytotoxicity in AML cell lines irrespective of multidrug-resistant status or karyotype.35 This agent was evaluated in a phase I study of R/R CD33+ AML and demonstrated blast clearance in 48% of the 85 evaluable patients, with CR/CRi in 27% (23 of 85), of whom 73% (of 23 patients) were negative for minimal residual disease among patients treated at the recommended dose of 40 µg/kg.36 Efficacy was even higher in the treatment-naïve patients (n = 27) with CR/CRi in 54% (14 of 27 evaluable patients).37 The most common grade 3 adverse events (AEs), occurring in 20% of patients, were neutropenia, thrombocytopenia, and anemia. Pre-clinical and early clinical-phase studies have shown SGN-CD33A to synergize with hypomethylating agents like azacitidine, and in a recently reported phase II study, the combination produced a CR/CRi rate of >60% with an 8-week mortality of 5% in frontline older patients with AML.38,39 This agent is currently being studied as monotherapy in maintenance; in the pre- and postallogeneic stem cell transplant (ASCT) settings; and in a phase III randomized study of SGN-CD33A with azacitidine versus azacitidine alone in untreated older patients with AML (NCT02326584, NCT02785900, NCT02706899, NCT02614560).
Another conjugated CD33 antibody that has shown marked activity both in vitro and in vivo is IMGN779.40,41 Its activity appears to be more selective to leukemic stem cells while sparing normal hematopoietic stem cells (HSCs), suggesting a potential for reduced myelosuppression. It is currently in a phase Ib trial in patients with relapsed AML (NCT02674763).
CD-123
Overexpression of the interleukin-3 (IL-3) receptor α-chain (IL-3 Rα/ CD123) on AML cells is associated with enhanced blast proliferation, disease relapse, and drug resistance in AML.42,43 Clinical activity was modest with the first-in-class anti-CD123 antibody (CSL360).44 Two second-generation antibodies (CSL362, SL-401) are being evaluated in phase I/II clinical trials. CSL362, a fully humanized antibody with a modified Fc-domain to enhance NK cell binding, was evaluated as postremission treatment in a phase I study involving 25 patients with CD123+ AML (in first or second CR), and it was able to prolong CRs beyond 6 months (26-52 weeks) in 10 of 20 evaluable patients.45 SL- 401 (DT388IL3), a human IL-3 ligand fused to a truncated diphtheria toxin,46 was evaluated in a phase I trial in 74 R/R or high-risk de novo AML patients, and it produced an ORR (CR + PR + disease stabilization) of 27% (20 of 74) and CR/CRi rate of 2.8% (2 of 74).The median survival of second-salvage AML patients treated in this trial (n = 33) was 3.2 months (range, 2-8.4), with 22% alive at 1 year.47 This compound is currently being evaluated in 3 phase II trials in hemato- logical malignancies including 1) in patients with relapsed AML and relapsed/frontline blastic plasmacytoid dendritic cell neoplasm; 2) maintenance after completion of consolidation in patients with high- risk AML patients who are not candidates for or have refused ASCT; and 3) in chronic myeloid disorders (NCT02113982, NCT02270463, and NCT02420873, respectively).
At this time, CD33- and CD123-targeted approaches are the most advanced. Monoclonal antibodies to other AML target anti- gens including CD25, CD37, and CD38 have recently begun clinical evaluation in phase I studies (NCT02588092, NCT02610062, and NCT01084252, respectively).
Radioimmunoconjugates
The radiosensitive nature of AML and its systemic pattern of involvement provide a rationale for radioimmunotherapy via targeted radionuclide antibody conjugates.48 Early studies with 2 beta-emitters, 131I-M195 and 131I-HuM195, demonstrated activity in myeloid leukemia, and demonstrated safety when used at myeloablative doses in conjunction with standard chemotherapeutics as a conditioning regimen for HSC transplantation.49,50 Subsequent clinical studies exploring antibodies against AML targets (CD33, CD45, or CD66) with beta emitters (131iodine, 188rhenium, 90yttrium), alone or as part of a conditioning regimen pre-ASCT in patients with relapsed AML, have demonstrated tolerability and encouraging clinical efficacy.51,52 The 131I-labeled anti-CD45 antibody BC8 (Iomab B) is currently being tested in phase I (NCT00589316) and phase III (NCT02665065) studies to evaluate its efficacy and safety as a part of myeloablative conditioning regimen prior to ASCT in patients with R/R AML. The properties of high-linear energy transfer and short particle length of decay has been exploited in the development of alpha emitter radioisotope conjugates. A phase I trial with the second-generation actinium- 225-lintuzumab (anti-CD33) antibody demonstrated reduction of bone marrow blasts in 65% of 18 evaluable patients with R/R AML.53 A phase I/II trial to determine the toxicity and efficacy of fractionated dosing of this agent in combination with LDAC in older untreated AML patients is ongoing (NCT02575963).
Immunotherapy in Acute Myeloid Leukemia
Immune Checkpoint Antibodies
The concept of targeting the immune system and not the tumor itself was initially achieved through monoclonal antibody-based inhibition of CTLA-4, a protein receptor on T cells that prevents them from unleashing an immune attack against tumor cells.
Another major approach to immune checkpoint blockade involves the inhibition of PD-1/PD-1 ligand, a co-inhibitory receptor- ligand system expressed on activated T cells and B cells.54 Clinical trials with antibodies targeting these pathways have demonstrated marked efficacy resulting in FDA approvals in a variety of solid tumors.55-57 Immune checkpoint therapy has more recently been evaluated in hematologic malignancies, with robust activity and approval in Hodgkin lymphoma and modest but clear activity in certain subsets of diffuse large B-cell lymphoma and in mantle cell lymphoma. The rationale to evaluate these agents in leukemia springs from studies demonstrating reduced AML burden and improved survival in murine models on inhibition of PD-1 and CTLA- 4.58-62 Additionally, PD-1 and other clinically targetable stimulatory checkpoint receptors, such as OX40 and ICOS, are overexpressed in the bone marrow of patients with relapsed AML.63
A phase I study with a PD-1/PD-L1 inhibitor, pidilizumab, demonstrated a minimal response in the form of stable disease in 1 of 8 salvage AML patients.64 DNA methyltransferase (DNMT) inhibitors increase expression of PD-1, PD-L1, and PD-L2 in patients with AML/myelodysplastic syndrome (MDS), with higher expression in DNMT inhibitor-resistant as compared with inhibitor-sensitive patients, suggesting that PD-1 upregulation may be involved in resistance to DNMT inhibitors.65 This has resulted in clinical trials combining PD-1/PD-L1 inhibitors with azacitidine in AML and MDS. A phase Ib/II study of nivolumab in combination with azacitidine in 53 patients with R/R AML (median prior therapies = 2, poor-risk cytogenetics = 45%) showed an ORR (CR, CRi, hematologic improvement) of 34%, and an 8-week mortality rate of 8%. The overall survival with this combination in the first salvage setting was superior to historical outcomes with other hypomethylating agent (HMA)-based salvage protocols at the same institution (9.3 vs 3.9 months, P = .03).66 Further- more, responses were durable, with 80% of relapsed patients who achieved CR/CRi alive at 1 year. PD-1 inhibitors are also being evaluated as a maintenance therapy, post induction and consolidation, in patients with high-risk AML who are not candidates for ASCT (NCT02532231) and in combination with standard induction chemotherapy in newly diagnosed younger (aged 18–60 years) patients with AML (NCT02464657). PD-1 inhibitors are also being evaluated in phase 1 trials for patients with AML after ASCT with initial encouraging results, with CR in 5 of 12 patients with post–SCT relapsed AML (median: 3 prior therapies before ASCT), including resolution of extramedullary leukemia in 3 of the patients (NCT01822509). Activation of stimulating check- points such as OX40 and ICOS represents another approach to enhance anti-tumor cytotoxicity, and this may prove synergistic in combination with checkpoint inhibition. Clinical trials with such inhibitory and stimulatory combinations are being planned.
Adoptive T-Cell Therapy
The process of adoptive cell transfer involves removal of T cells from a patient and modifying them so that they express receptors specific to the patient’s particular cancer. The ex vivo expanded cytotoxic T lymphocytes, now capable of targeting tumor-associated antigens, are reinfused into patients. The genetically engineered receptors consist of an extracellular domain created by the fusion between the variable region of heavy and light chains of an antigen-specific monoclonal antibody and of an intracellular T cell-activating domain, usually CD3-ζ. The tumor specificity of the monoclonal antibodies allows activation of CAR-T cells independent of major histocompatibility complex (MHC). After showing success in acute lymphoblastic leukemia, the approach was recently evaluated in the treatment of patients with AML67,68 with CAR-T cells targeted to Ley, a carbohydrate antigen that is overexpressed by malignant myeloid cells. The pilot study (n = 5) involved 4 evaluable patients with relapsed AML, 2 of whom had achieved stable disease and 2 who had transient responses.69 The investigators observed that Ley was only expressed in a proportion of AML blasts, unlike epithelial tumors, in which the expression was more uniform and intense. Therefore, there has been further development of this agent in lung cancer therapy only. Preclinical studies have shown that anti-CD123 CAR-Ts are capable of targeting AML blasts and leukemia stem cells, suggesting these could be promising agents for AML CAR-T cell approaches.70 A number of phase I clinical trials including anti-CD33, -CD7, and -CD133 CAR-Ts (NCT01864902, NCT02799680, NCT02742727, NCT02541370) and anti-CD123 and anti-NKG2D ligand CAR-T cells (NCT02159495, NCT02623582; NCT02203825) studies are ongoing in patients with R/R AML. The role and future of CAR-T cell therapy in AML remains unknown at this time.
T Cell-Engaging Antibodies
BiTE antibodies are fusion protein constructs consisting of 2 single-chain variable fragments of 2 antibodies; one binds to T cells via the CD3 receptor, and the other binds to a tumor cell via a tumor-specific antigen. BiTE antibodies effectively recruit T cells and link them with tumor cells, thereby effectuating T cell-mediated cytotoxic activity on tumor cells independent of the presence of MHC-I or costimulatory molecules.71,72 After the promising clinical activity and FDA approval of blinatumomab, an anti-CD19/CD3 first-in-class BiTE antibody in precursor B-cell acute lymphoblastic leukemia, the approach has been adapted to target AML with the development of novel constructs targeting anti-CD3/CD33, such as AMG 330.73,74 Preclinical studies have demonstrated potent activity of this agent in CD33+ AML74-76 and a phase I trial of AMG330 is ongoing (NCT02520427). Phase I trials evaluating XmAb CD3/CD123 BiTE (NCT02730312) and JNJ-63709178 CD123/CD3 (NCT02715011) have recently begun enrollment. Early data suggest that close monitoring for cytokine release syndrome will be important with such bispecific approaches in AML.
Next-Generation Hypomethylating Agents
Guadecitabine (SGI-110), a second-generation hypomethylating agent, improves upon the pharmacokinetics of decitabine by incorporating deoxyguanosine dinucleotide into decitabine, thereby increasing in vivo exposure and potentially improving efficacy.77 The outcomes of a phase II study involving 103 patients with R/R AML treated with SGI-110 were recently presented. CR and median survival rates on the 10-day (n = 53) and 5-day (n = 50) regimens were 30% and 16% at 7.1 and 5.7 months, respectively; the most common grade 3 AEs were anemia, thrombocytopenia, neutropenia, pneumonia, and sepsis.78 A phase III trial comparing SGI-110 with standard of care in previously treated AML is currently ongoing (NCT02920008).
Other Therapies
DOT1L-like/histone H3K79 methyltransferase/DOT1L is a protein implicated in the development of mixed lineage leukemia (MLL) through aberrant hypermethylation-induced gene expression. A phase I trial evaluating pinometostat (EPZ-5676) in adult R/R MLL (n = 49) determined the agent to have clinical activity (composite CR [n = 2], PR [n = 1], leukemia cutis [n = 3]) with an acceptable safety profile (NCT01684150).79 Another promising approach involves targeted inhibition of chromatin regulatory proteins such as lysine-specific demethylase-1 (LSD1), an enzyme responsible for histone H3 demethylation apart from other functions. GSK2879552 is an orally administered LSD1 inhibitor that is currently undergoing development in a phase I dose-escalation study involving R/R AML (NCT02177812), and results are eagerly awaited.
The bromodomain (BRD) family is an epigenetic class of his- tone-modification proteins with an ability to “read” the genome and modulate gene expression through transcriptional regulator recruitment to specific genome locations. BRDs of these reader proteins promote aberrant gene expression and sustain leukemic maintenance, thus providing a rationale for developing inhibitors against this class.80 Berthon and colleagues reported on outcomes with OTX105, an oral BRD inhibitor, in a phase I dose-escalation study.81 Among 36 patients with R/R AML, 3 achieved CR/CRi, and 2 had partial blast clearance. Recommended dose for further phase II studies was 80 mg on a schedule of 14 days on, 7 days off.
Histone deacetylase inhibitors provide another therapeutic approach to exploit the aberrant epigenetic alterations in AML. After showing single-agent activity in a phase I trial in AML, pracinostat was evaluated in combination with azacitidine in a phase II study involving older patients with AML deemed to not be candidates for intensive chemotherapy.82 The combination arm proved superior, with an estimated median OS of 19.1 months, resulting in the FDA granting a breakthrough designation for the combination’s use in elderly patients with AML aged ≥75 years, or ineligible for intensive chemotherapy.
Pevonedistat is a novel NEDD8-activating enzyme inhibitor with single-agent clinical activity in R/R AML.83 Swords and colleagues presented outcomes of a phase II study evaluating pevonedistat in combination with azacitidine in treatment-naïve older patients with AML.84 Among 61 patients treated, ORRs were observed in 52 (18 CR, 5 CRi, 8 PR, 21 other) with a median duration of re- mission of 8.3 months. After a median follow-up of 16.4 months, projected 6-month survival was 52%. Single-agent and hypomethylating combination strategies with pevonedistat and MLN4924, another inhibitor in this class, are ongoing (NCT03009240, NCT02610777, and NCT01814826). NCT00911066 is completed.Conclusion
Among available molecular targeted therapies, FLT3 inhibitors, as single agents or in combination with standard chemotherapy, in frontline or salvage settings, have already shown benefit (frontline 7+3 with midostaurin in untreated young AML patients in a phase III trial) or are being evaluated in phase III trials. Other inhibitors targeting IDH1/2 and BCL-2 among other small molecule inhibitors have shown promise as single agents and in combination with DNMT inhibitors, and are being evaluated in ongoing expanded phase II trials or soon-to-open phase III trials. Monoclonal antibody conjugates, such as SGN-CD33A and GO, are currently being evaluated in combination strategies with DNMT inhibitors or in cytotoxic induction regimens in phase III trials, while other immune- and antibody-based therapies, discussed above, are still in early phases of clinical development. The integration of informative biomarkers into clinical practice, and trials and implementation of rational combinatorial strategies of targeted, immune, monoclonal, and cytotoxic chemotherapies with each other, all while assessing for tolerability and toxicity, are important steps forward to help define and expand the scope of these novel therapies in AML.
Affiliations: All authors are with the Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, Texas.
Send correspondence to: Naval Daver, MD, Department of Leukemia, The University of Texas M.D. Anderson Cancer Center, 1515 Holcombe Blvd, Unit 428, Houston, TX 77030. E-mail: [email protected].
Funding source: This manuscript was supported in part by the MD Anderson Cancer Center Leukemia Support Grant CA016672 and by generous philanthropic contributions to the M.D. Anderson Moon Shots Program.
Disclosures: The authors report no relationship or financial interest with any entity that would pose a conflict of interest with the subject matter of this article.
References
- Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854-4860. doi: 10.1182/blood-2013-01-466706.
- Garcia-Manero G, Othus M, Pagel JM, et al. A randomized phase III study of standard cytarabine plus daunorubicin (7+3) therapy versus idarubicin with high dose cytarabine (IA) with or without vorinostat (IA+V) in younger patients with previously untreated acute myeloid leukemia (AML). Paper presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 901. https://ash.confex.com/ash/2016/webprogram/Session8689. html. Accessed February 17, 2017.
- Grimwade D, Ivey A, Huntly BJP. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29-41. doi: 10.1182/blood-2015-07-604496.
- Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33(11):563-570. doi: 10.1016/j.it.2012.06.002.
- Martelli MP, Gionfriddo I, Mezzasoma F, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125(22):3455-3465. doi: 10.1182/blood-2014-11-611459.
- Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106-1117.
- Daver N, Cortes J, Kantarjian H, Ravandi F. Acute myeloid leukemia: advancing clinical trials and promising therapeutics. Expert Rev Hematol. 2016;9(5):433-445. doi: 10.1586/17474086.2016.1158096.
- Röllig C, Serve H, Hüttmann A, et al; Study Alliance Leukaemia. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691-1699. doi: 10.1016/ S1470-2045(15)00362-9.
- Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655-4662. doi: 10.1182/blood-2013-01-480228.
- Hills RK, Gammon G, Trone D, Burnett AK. Quizartinib significantly improves overall survival in FLT3-ITD positive AML patients relapsed after stem cell transplantation or after failure of salvage chemotherapy: a comparison with historical AML database (UK NCRI data). Blood. 2015;126(23):2557-2557.
- Abdelall W, Kantarjian HM, Borthakur G, et al. The combi- nation of quizartinib with azacitidine or low dose cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: interim report of a phase I/II trial. Paper presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 1642. https://ash.confex.com/ash/2016/webprogram/Paper94012. html. Accessed February 17, 2017.
- Perl AE, Altman JK, Cortes JE, et al. Final results of the Chrysalis Trial: a first-in-human phase 1/2 dose-escalation, dose-expansion study of gilteritinib (ASP2215) in patients with relapsed/refractory acute myeloid leukemia (R/R AML). Paper presented at: 58th Annual Meeting & Exposition of the Amer- ican Society of Hematology; December 3-6, 2016; San Diego, CA. http://www.bloodjournal.org/content/128/22/1069?s- so-checked=true. Accessed February 17, 2017.
- Stone RM, Dohner H, Ehninger G, et al. CALGB 10603 (RATIFY): A randomized phase III study of induction (daunorubi- cin/cytarabine) and consolidation (high-dose cytarabine) chemo- therapy combined with midostaurin or placebo in treatment-naïve patients with FLT3 mutated AML. 2011 ASCO Annual Meeting. J Clin Oncol. 2011;29 (suppl; abstr TPS199).
- Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood. 2014;124(21):389-389.
- Wang ES, Stone RM, Tallman MS, et al. Crenolanib, a type I FLT3 TKI, can be safely combined with cytarabine and anthracycline induction chemotherapy and results in high response rates in patients with newly diagnosed FLT3 mutant acute myeloid leukemia (AML). Blood. 2016;128(22):1071.
- Knapper S, Burnett AK, Littlewood T, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108(10):3262-3270. doi: https://doi.org/10.1182/blood-2006-04-015560.
- Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117: 3294-3301. doi: 10.1182/blood-2010-08-301796.
- DiNardo CD, Schimmer AD, Yee KWL, et al. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 1073. https://ash.confex.com/ash/2016/webpro- gram/Paper92606.html. Accessed February 17, 2017.
- Agios announces new clinical data from dose-escalation portion of phase 1 trial of single agent AG-120 showing durable molecular responses in patients with advanced hematologic malignancies [news release]. San Diego, CA: Agios Pharmaceuticals, Inc; December 5, 2016. http://investor.agios.com/phoenix. zhtml?c=251862&p=irol-newsArticle&ID=2227727. Accessed on March 17, 2017.
- DiNardo C, Pollyea D, Pratz K, et al. A phase 1b study of venetoclax (ABT-199/GDC-0199) in combination with decitabine or azacitidine in treatment-naïve patients with acute myelogenous leukemia who are ≥ to 65 years and not eligible for standard induction therapy. Blood. 2015;126(23):327.
- Wei A, Strickland SA, Roboz GJ, et al. Safety and efficacy of venetoclax plus low-dose cytarabine in treatment-naïve patients aged ≥65 years with acute myeloid leukemia. Paper presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 102. https://ash.confex.com/ash/2016/webprogram/Paper91423. html. Accessed March 17, 2017.
- Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML). Blood. 2003;101(8):3142-3149. doi: https://doi.org/10.1182/ blood-2002-10-3062.
- Saito Y, Kitamura H, Hijikata A, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2(17):17ra9. doi: 10.1126/ scitranslmed.3000349.
- van Rhenen A, van Dongen GAMS, Kelder A, et al. The novel AML stem cell-associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659- 2666. doi: https://doi.org/10.1182/blood-2007-03-083048.
- Shuptrine CW, Surana R, Weiner LM. Monoclonal antibodies for the treatment of cancer. Semin Cancer Biol. 2012;22(1):3-13. doi: 10.1016/j.semcancer.2011.12.009.
- Lee EM, Lock RB. Targeting of AML-leukemic stem cells with monoclonal antibodies. Future Oncol. 2009;5(9):1327-1330. doi: 10.2217/fon.09.115.
- Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490-1496.
- Larson RA, Sievers EL, Stadtmauer EA, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104(7):1442-1452.
- Sievers EL, Larson RA, Stadtmauer EA, et al; Mylotarg Study Group. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19(13):3244-3254.
- Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369-377. doi: 10.1200/ JCO.2010.31.4310.
- Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012;30(32):3924-3931. doi: 10.1200/JCO.2012.42.2964.
- Castaigne S, Pautas C, Terré C, et al; Acute Leukemia French Association. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508-1516. doi: 10.1016/S0140-6736(12)60485-1.
- Perini GF, Santos FP, Esteves I, et al. Use of gemtuzumab ozogamycin combined with conventional chemotherapy in patients with acute myeloid leukemia. Einstein (Sao Paulo). 2011;9(2):190- 195. doi: 10.1590/S1679-45082011AO1987.
- Burnett AK, Hills RK, Hunter AE, et al; UK National Cancer Research Institute AML Working Group. The addition of gemtuzumab ozogamicin to low-dose Ara-C improves remission rate but does not significantly prolong survival in older patients with acute myeloid leukaemia: results from the LRF AML14 and NCRI AML16 pick-a-winner comparison. Leukemia. 2013;27(1):75-81. doi: 10.1038/leu.2012.229.
- Kung Sutherland MS, Walter RB, Jeffrey SC, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122(8):1455-1463. doi: 10.1182/blood-2013-03-491506.
- Stein AS, Walter RB, Erba HP, et al. A phase 1 trial of SGN- CD33A as monotherapy in patients with CD33-positive acute myeloid leukemia (AML). Blood. 2015;126(23):324.
- Bixby DL, Fathi AT, Kovacsovics TJ, et al. Vadastuximab talirine monotherapy in older patients with treatment naïve CD33-positive acute myeloid leukemia. Presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 590. https://ash. confex.com/ash/2016/webprogram/Paper90715.html. Accessed February 17, 2017.
- Sutherland MK, Yu C, Anderson M, et al. 5-azacytidine enhances the anti-leukemic activity of lintuzumab (SGN-33) in preclinical models of acute myeloid leukemia. MAbs. 2010;2(4):440-448.
- Fathi AT, Erba HP, Lancet JE, et al. SGN-CD33A plus hypomethylating agents: a novel, well-tolerated regimen with high remission rate in frontline unfit AML. Blood. 2015;126(23):454.
- Whiteman KR, Noordhuis P, Walker R, et al. The anti- body-drug conjugate (ADC) IMGN779 is highly active in vitro and in vivo against acute myeloid leukemia (AML) with FLT3-ITD mutations. Blood. 2014;124(21):2321.
- Krystal WM, Walker R, Fishkin N, et al. IMGN779, a CD33-targeted antibody-drug conjugate (ADC) with a novel DNA-alkylating effector molecule, induces DNA damage, cell cycle arrest, and apoptosis in AML cells. Blood. 2015;126(23):1366.
- Bagley CJ, Woodcock JM, Stomski FC, Lopez AF. The structural and functional basis of cytokine receptor activation: lessons from the common beta subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood. 1997;89(5):1471-1482.
-
Testa U, Riccioni R, Militi S, et al. Elevated expression of
IL-3R alpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980-2988. doi: https://doi.org/10.1182/ blood-2002-03-0852. - He SZ, Busfield S, Ritchie DS, et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma. 2015;56(5):1406-1415. doi: 10.3109/10428194.2014.956316.
- Smith BD, Roboz GJ, Walter RB, et al. First-in-man, phase 1 study of CSL362 (anti-IL3Rα / anti-CD123 monoclonal antibody) in patients with CD123+ acute myeloid leukemia (AML) in CR at high risk for early relapse. Blood. 2014;124(21):120.
- Frankel AE, Ramage J, Kiser M, et al. Characterization of diphtheria fusion proteins targeted to the human interleukin-3 receptor. Protein Eng. 2000;13(8):575-581.
- Konopleva M, Hogge DE, Rizzieri DA, et al. Phase I trial results for SL-401, a novel cancer stem cell (CSC) targeting agent, demonstrate clinical efficacy at tolerable doses in patients with heavily pre-treated AML, poor risk elderly AML, and high risk MDS. Blood. 2010;116(21):3298.
- Clift RA, Buckner CD, Appelbaum FR, et al. Allogeneic marrow transplantation in patients with acute myeloid leukemia in first remission: a randomized trial of two irradiation regimens. Blood. 1990;76(9):1867-1871.
- Jurcic JG, Caron PC, Nikula TK, et al. Radiolabeled an- ti-CD33 monoclonal antibody M195 for myeloid leukemias. Cancer Res. 1995;55(23 Suppl):5908s-5910s.
- Feldman E, Kalaycio M, Weiner G, et al. Treatment of relapsed or refractory acute myeloid leukemia with humanized anti-CD33 monoclonal antibody HuM195. Leukemia. 2003;17(2):314-318.
- Pagel JM, Appelbaum FR, Eary JF, et al. 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood. 2006;107(5):2184-2191. doi: https://doi.org/10.1182/blood-2005-06-2317.
- Bunjes D, Buchmann I, Duncker C, et al. Rhenium 188-la- beled anti-CD66 (a, b, c, e) monoclonal antibody to intensify the conditioning regimen prior to stem cell transplantation for patients with high-risk acute myeloid leukemia or myelodysplastic syndrome: results of a phase I-II study. Blood. 2001;98(3):565-572. doi: https://doi.org/10.1182/blood.V98.3.565.
- Jurcic JG, Ravandi F, Pagel JM, et al. Phase I trial of targeted alpha-particle therapy using actinium-225 (225Ac)-lintuzumab (anti-CD33) in combination with low-dose cytarabine (LDAC) for older patients with untreated acute myeloid leukemia. Blood. 2014;124(21):5293.
- Sui X, Ma J, Han W, et al. The anticancer immune response of anti-PD-1/PD-L1 and the genetic determinants of response to anti-PD-1/PD-L1 antibodies in cancer patients. Oncotarget. 2015;6(23):19393-19404. Review.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723. doi: 10.1056/NEJMoa1003466.
- Page DB, Postow MA, Callahan MK, et al. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185-202. doi: 10.1146/annurev-med-092012-112807.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517-2526. doi: 10.1056/NEJMoa1104621.
- Mumprecht S, Schürch C, Schwaller J, et al. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114(8):1528-1536. doi: 10.1182/blood-2008-09-179697.
- Fevery S, Billiau AD, Sprangers B, et al. CTLA-4 blockade in murine bone marrow chimeras induces a host-derived antileukemic effect without graft-versus-host disease. Leukemia. 2007;21(7):1451-1459.
- Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114(8):1545-1552. doi: 10.1182/ blood-2009-03-206672.
- Zhou Q, Munger ME, Highfill SL, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116(14):2484-2493. doi: 10.1182/ blood-2010-03-275446.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734-1736.
- Daver N, Basu S, Garcia-Manero G, et al. Defining the immune checkpoint landscape in patients (pts) with acute myeloid leukemia. Presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. http://www.bloodjournal.org/content/128/22/2900?s- so-checked=true. Accessed February 17, 2017.
- Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14(10):3044-3051. doi: 10.1158/1078- 0432.CCR-07-4079.
- Yang H, Bueso-Ramos C, DiNardo C, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280-1288. doi: 10.1038/leu.2013.355.
- 66. Daver N, Basu S, Garcia-Manero G, et al. Phase IB/II study of nivolumab in combination with azacytidine (AZA) in patients (pts) with relapsed acute myeloid leukemia (AML). Presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 763. https://ash.confex.com/ash/2016/webprogram/Paper98062. html. Accessed February 17, 2017.
- Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509-1518. doi: 10.1056/NEJMoa1215134.
- Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemo- therapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138. doi: 10.1126/scitranslmed.3005930.
- Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122-2129. doi: 10.1038/mt.2013.154.
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28(8):1596-1605. doi: 10.1038/leu.2014.62.
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69(12):4941-4944. doi: 10.1158/0008-5472.CAN-09-0547.
- Nagorsen D, Bargou R, Ruttinger D, et al. Immunotherapy of lymphoma and leukemia with T-cell engaging BiTE anti- body blinatumomab. Leuk Lymphoma. 2009;50(6):886-891. doi: 10.1080/10428190902943077.
- Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493-2498. doi: 10.1200/JCO.2010.32.7270.
- Aigner M, Feulner J, Schaffer S, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27(5):1107-1115.
- Laszlo GS, Gudgeon CJ, Harrington KH, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554-561. doi: 10.1182/blood-2013-09-527044.
- Harrington KH, Gudgeon CJ, Laszlo GS, et al. The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, AMG 330, is impacted by disease stage and risk. PLoS One. 2015;10(8):e0135945. doi: 10.1371/journal.pone.0135945.
- Issa JP, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16(9):1099-1110. doi: 10.1016/S1470-2045(15)00038-8.
- Daver N, Kantarjiam HM, Roboz GJ, et al. Long term survival and clinical complete responses of various prognostic subgroups in 103 relapsed/refractory acute myeloid leukemia (r/r AML) patients treated with guadecitabine (SGI-110) in phase 2 studies. Presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 904. https://ash.confex.com/ash/2016/webprogram/ Paper91389.html. Accessed February 17, 2017.
- Stein EM, Garcia-Manero G, Rizzieri DA, et al. A phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in adults with relapsed or refractory leukemia: safety, clinical activity, expo- sure and target inhibition. Blood. 2015;126(23):2547.
- Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33(3):146-153. doi: 10.1016/j.tips.2011.12.002.
- Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3(4):e186-e195. doi: 10.1016/ S2352-3026(15)00247-1.
- Garcia-Manero G, Atallah E, Odenike O, et al. Pracinostat in combination with azacitidine produces a high rate and rapid onset of disease remission in patients with previously untreated acute myeloid leukemia (AML). Blood. 2014;124(21):947.
- Swords RT, Erba HP, DeAngelo DJ, et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169(4):534-543. doi: 10.1111/bjh.13323.
- Swords RT, Coutre S, Maris MB, et al. Results of a clinical study of pevonedistat (Pev), a first-in-class NEDD8-activating enzyme (NAE) inhibitor, combined with azacitidine (Aza) in older patients (Pts) with acute myeloid leukemia (AML). Paper presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 98. https://ash.confex.com/ash/2016/webprogram/Paper93300. html. Accessed March 21, 2017.
- Cortes JE, Perl AE, Dombret H, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥ 60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. Blood. 2012;120(21):48.
- Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-con- trolled trial. J Clin Oncol. 2013;31(25):3110-3118. doi: 10.1200/ JCO.2012.46.4990.
- Stone RM, Mandrekar S, Sanford BL, et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C) induction (ind), high-dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18-60 with FLT3 mutations (muts): an international prospective randomized (rand) p-con- trolled double-blind trial (CALGB 10603/RATIFY [Alliance]). Blood. 2015;126(23):6.
- Stein EM, DiNardo C, Altman JK, et al. Safety and efficacy of AG-221, a potent inhibitor of mutant IDH2 that promotes differentiation of myeloid cells in patients with advanced hematologic malignancies: results of a phase 1/2 trial. Blood. 2015;126(23):323.
- Agios announces new data from ongoing phase 1 trial of AG-120 showing durable clinical activity in patients with advanced hematologic malignancies [news release]. Cambridge, MA: Agios Pharmaceuticals, Inc; June 12, 2015. http://inves- tor.agios.com/phoenix.zhtml?c=251862&p=RssLanding&- cat=news&id=2058807. Accessed on March 21, 2017.
- DiNardo CD, Schimmer AD, Yee KWL, et al. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. 2016;128(22):1073.
- Andreeff M, Kelly KR, Yee K, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Can- cer Res. 2016;22(4):868-876. doi: 10.1158/1078-0432.CCR-15-0481.
- Erba HP, Levy MY, Vasu S, et al. A phase 1b study of vadastuximab talirine in combination with 7+3 induction therapy for patients with newly diagnosed acute myeloid leukemia (AML). Presented at: 58th Annual Meeting & Exposition of the American Society of Hematology; December 3-6, 2016; San Diego, CA. Abstract 211. https://ash.confex.com/ash/2016/webprogram/ Paper90718.html. Accessed March 21, 2017.