Introduction
Over the past 5 years, immunotherapy has brought a revolution of changes in how numerous cancers are treated. Using the immune system to target tumors, largely by suppressing the programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte associated protein 4 (CTLA-4) pathways, has been effective for melanoma, non–small cell lung cancer, head and neck cancer, renal cell carcinoma, and Hodgkin’s lymphoma.1-5 Although PD-1 and CTLA-4 inhibitors represent significant advances in treatment and, in many cases, durable remissions, response rates have ranged between 10% and 61%, leaving many patients needing alternative therapy. Advances in the understanding of the interactions between tumors and the immune system are leading to even more novel cancer immunotherapeutics.
Background
Progression from a contained neoplasm to a metastatic state requires successfully evading the immune system.6 Tumors have complex mechanisms by which they alter antigen processing and presentation within their microenvironments, as well as the activation process of an immune response that shields the tumor from immune surveillance and suppresses surrounding leukocytes.Leukocytes infiltrating the tumor microenvironment are suppressed in their immune function and used by the neoplasm to harness growth factors for proliferation, angiogenesis, and pro- longed survival of the tumor cells. Tumor-infiltrating macrophages (TAMs), for example, are recruited to the tumor with chemokines, such as C-C motif ligand 2 (CCL2), then experience prolonged survival with cytokines, such as vascular endothelial growth factor (VEGF) and colony-stimulating factors within the tumor microenvironment.7 TAMs then produce growth factors and interleukins leading to tumor proliferation, VEGF and fibroblast growth factor 2 leading to angiogenesis, and chemokines leading to adaptive immune suppression.7
Inhibiting an adaptive T-cell immune response against the tumor is also mediated by a complex mechanism. TAMs and myeloid- derived suppressor cells (MDSCs) in the tumor microenvironment produce suppressive indoleamine dioxygenase metabolites and tumor necrotic factors.8-10 Chemokines, such as interleukin 10 (IL- 10), transforming growth factor-beta (TGF-beta), and macrophage colony-stimulating factor, in the tumor microenvironment inhibit dendritic cell maturation leading to impaired antigen presentation and T-cell energy.6,7 Regulatory T cells (Tregs), which suppress T-cell activation through TGF-beta and IL-10, are recruited to the tumor microenvironment, partially by the chemokine CCL2 from TAMs.6
Finally, the overexpression of immune checkpoint inhibitors on various cells within the microenvironment comprises a multi- factorial suppressive signal to prevent a strong T-cell–mediated response. Some of these pathways, particularly CTLA-4 and PD-1, have already been used with success in the clinical setting. CTLA-4 is a cell membrane protein receptor expressed on T cells, particularly Tregs, which binds to CD80 and CD86 on antigen-presenting cells (APCs) and effectively prevents the APCs from binding to CD28 to T cells and triggering an immune response as depicted in Figure 1. PD-1 is another surface protein on T cells that upon binding to its ligand, programmed cell death ligand (PD-L1), inhibits T-cell activation and promotes apoptosis in antigen-specific T cells.11 Inhibitors against CTLA-4 and PD-1 have been very successful clinically, and have become the standard of care for several cancers. Other checkpoints—such as OX40, LAG-3, TIGIT, TIM-3, and BLTA—are being investigated as other potential immunotherapeutic targets (Figure 2).
Understanding the tumor microenvironment and how it interacts with the immune system represent potential breakthroughs for therapeutic targets and implications. The next directions for immune therapy include mechanisms to increase innate activation of antitumor T cells, altering the tumor microenvironment that will confer immunogenicity to the tumor, engineering an antitumor immune response through adoptive T-cell therapy, or inhibiting tumor-mediated immune suppression. Multiple vaccine trials are underway to establish an innate immune response against tumors, and drugs inhibiting checkpoint pathways or abrogating signals of TAMS, MDSCs, and Tregs are in development toward these goals.
Activating the Immune Response
Increasing the tumor-specific immune response by using agonist antibodies targeting activating checkpoints, or by increasing the antigen presentation process to T cells through vaccines and oncolytic viruses is a potential direction of further therapeutic studies.
Agonistic Checkpoints
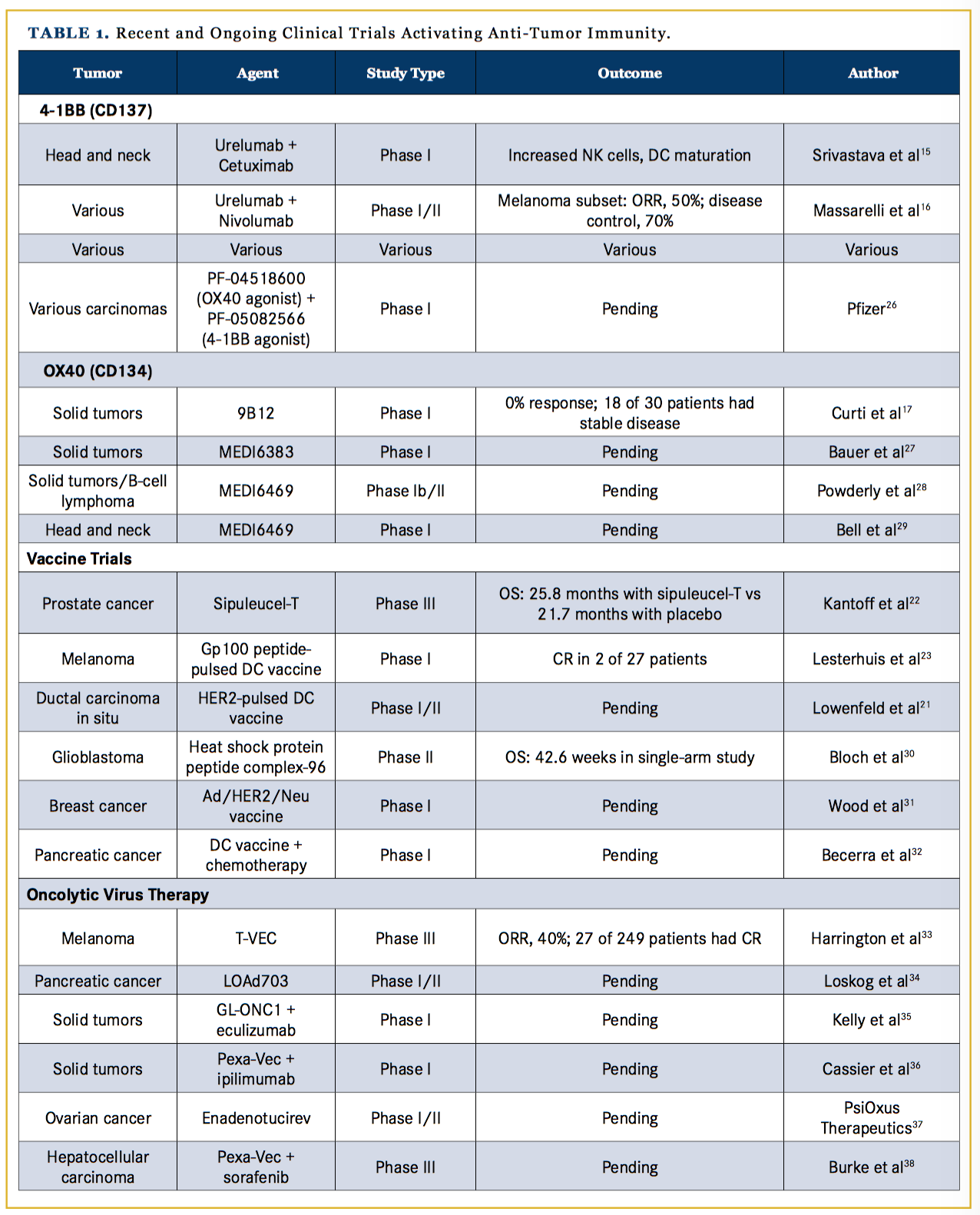
Activating checkpoints, such as CD137 and OX40, within the tumor microenvironment could be targets of agonistic agents used to activate an immune response against the tumor. CD137, or 4-1BB, is a tumor necrotic factor receptor found primarily on activated T cells, natural killer (NK) cells, and myeloid cells. When CD137 binds its ligand, CD137L, which is found on APCs, such as dendritic cells and macrophages, the binding leads to a stimulatory signal to activate CD4+ and CD8+ T cells by in- creasing IL-2, IL-4, and interferon-gamma as outlined in Figure 1.12 CD137 antibodies have been effective in murine models at increasing antitumor immune responses while decreasing the humoral immune response and antibody production that leads to autoimmune diseases.13,14 A clinical phase I trial with urelumab, a humanized IgG CD137 antibody, has shown increased antitumor T-cell and NK cell responses in patients with head and neck cancer.15 A phase 1 trial of urelumab plus nivolumab showed an overall response rate (ORR) of 50% among 46 patients with metastatic melanoma, with no increase in treatment-related adverse events compared with nivolumab alone; response rates were notably statistically similar between patients with PD-1–positive and PD-1–negative tumors, and 6 patients with lymphoma treated with urelumab alone had a partial response (PR) or a complete response (CR).16
OX40 is a costimulatory transmembrane glycoprotein receptor on activated T cells. Ligands on APCs binding to OX40 induce nuclear factor kappa enhancer of activated B cells, which leads to increased T-cell proliferation, cytotoxic activity, and survival.17 While it is known that the binding of OX40 to its ligand has positive influences on cytotoxic T cells, OX40 activity on Tregs is not completely understood; along with mild stimulatory activity on Tregs, OX40 has been found to decrease Treg-mediated immune suppression although the mechanism is not well understood.18 OX40 antibodies in murine models with an intact immune system have demonstrated tumor regression. A phase I clinical trial in human patients showed no clinical responses by RECIST criteria, although 18 of 30 patients showed stable disease or regression in at least 1 lesion. OX40 antibody treatment was well tolerated with grade 3 and 4 toxicities related solely to lymphopenia.17 Currently, preclinical studies with combination therapy using OX40 with other targets are underway (Table 1).19,-22
Vaccines
Another mechanism to activate an antitumor T-cell response is through vaccines, especially those that target dendritic cells. The dendritic cells within tumors have been thought to be deficient in presenting antigens to T cells because of incomplete maturity; investigations with dendritic cell vaccines combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) or cyclic dinucleotides, which activate the stimulator of interferon gene complex (STING) pathway, trigger activation of dendritic cells.23,24
Whereas vaccines utilizing antigen-presenting dendritic cells to mount a T-cell response have been investigated for several decades in a wide variety of cancers, and have shown increased immunogenicity in murine models and cell cultures, their translation into a meaningful clinical response has been limited.25 One exception is the success of sipuleucel-T, a vaccine made with autologous monocytes pulsed with prostatic acid phosphatase antigens and GM-CSF ex vivo, and then injected into patients with metastatic prostate cancer. Sipuleucel-T led to a 4.1-month survival advantage in a phase III clinical trial compared with placebo.26
Currently, with an increasing understanding of tumor immunology, a wide range of uses for dendritic cell vaccines are being investigated, with clinical trials being run for adjuvant melanoma,27 neoadjuvant HER2+ ductal carcinoma in situ,28 and glioblastomas,29 among many others shown in Table 1.30-32 Notably, however, vaccines have been a challenging therapy to pursue because they take significant time to manufacture; thus, their use is limited to patients who are not at risk of progressing while they await their treatment (the typical wait time is 4-6 weeks), such as patients in the adjuvant setting. The combination of vaccines with checkpoint inhibitors is also under investigation.32
Viral Therapy
Oncolytic viruses are another class of agents used to incite an immune reaction against a tumor. Initially derived from herpes simplex virus (HSV), these viruses are engineered to specifically target cancer cells for replication and toxicity. HSV induces a strong immune response, including activation of innate cells (such as T cells and NK cells), and a humoral response by triggering cytokine cascades, complement proteins, and immunoglobulin. Oncolytic viruses are designed to harness immune responses against the tumor cells when they are injected into the tumor microenvironment.33
Talimogene laherparepvec (T-VEC) is the first oncolytic virus to be approved by the FDA for intralesional clinical use. In a phase III trial comparing T-VEC with GM-CSF, 436 patients with melanoma having unresectable stage IIIB, IIIC, and stage IV with an isolated site of metastasis, were randomized to GM-CSF or T-VEC. Among 249 patients treated with T-VEC, the ORR was 40% (compared with 2.3% with GM-CSF) and 27 patients had a CR.34 T-VEC was also studied with ipilimumab in a phase I trial of 19 patients with unresectable melanoma; the ORR was 50%, with an impressive 44% of patients having a CR and a median progression-free survival (PFS) of 18 months.35 Using T-VEC in combination with other checkpoint inhibitors, such as PD-1, is being evaluated in trials, including a phase III trial in melanoma.34,36
Malignant gliomas have also been an active area of research for oncolytic viral therapy. In a phase I trial of the modified HSV1 virus, G207, followed by radiation, 6 of 9 patients with malignant gliomas had stable disease or a partial response (PR), although no survival benefit was noted.37 A more successful recombinant virus, PVSRIPO, has been designated by the FDA as a break- through therapy for high-grade gliomas. PVSRIPO is made from a poliovirus, with the internal ribosomal entry site exchanged for that of the human rhinovirus 1 that abrogates the ability to replicate and damage neuronal cells; the virus is still able to enter and proliferate in glioma cells, but now it leads to an inflammatory reaction against the glioma cells.37 A phase I trial with PVSRIPO in grade 4 glioma patients showed a median OS of 12.6 months compared with 10.5 months in historic controls; significantly, 23% of patients were alive at 2 years compared with 10% in the control arm.37 Currently, adenoviruses, HSV, the measles virus, retroviruses, and parvoviruses are under investigation for potential oncolytic viral therapies.34,38-42
Innate Immunity
Generalized activation of the innate immune response through the STING pathway and toll-like receptors (TLRs) may also be used to induce an antitumor immune response. Injecting murine tumors with cyclic diguanylate monophosphate, an agonist of the STING pathway, improved the survival of mice with brain gliomas and showed increased T-cell infiltrates in the tumors.43 Similarly, systemically injecting mice with melanoma with the dendritic cell growth factor FLT3L, followed by intra-tumoral poly I:C injections and treatment with PD-1 or BRAF inhibition, showed improved responses compared with the PD-1 or BRAF inhibitors alone.44,45 Combining dendritic cell-activating agents with checkpoint inhibitors may expand and prolong responses to patients who do not respond to PD-1 or CTLA-4 inhibitors alone.
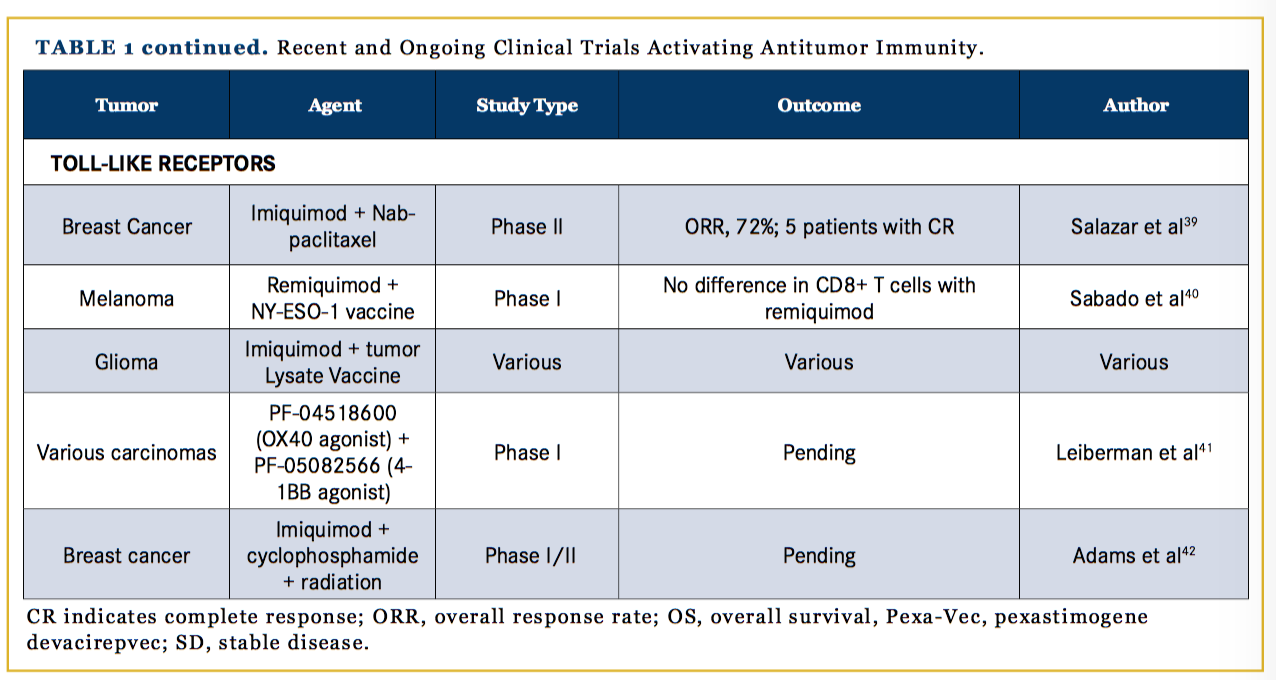
TLRs bind antigens and increase cytokine secretion that induce an immune response through activated Langerhans cells, macrophages, and lymphocytes; in T cells, TLRs stimulate proliferation via the protein kinase B pathway, as shown in Figure 1. TLR agonists have been shown to inhibit tumor growth in preclinical murine models by countering immune suppression in the tumor microenvironment.46 Imiquimod, the topical TLR agonist, is FDA-approved for cutaneous basal cell carcinoma, condylomata acuminata, and actinic keratosis. Clinical trials with imiquimod for other cutaneous applications, such as breast cancer with skin involvement, have been promising. In a phase II trial of 14 breast cancer patients, 5 had a CR and the ORR was 72%.47 In another trial, remiquimod, the topical TLR 7/8 agonist, was studied as an agonist with a cancer testis antigen vaccine for melanoma patients; the results of 20 patients showed no significant difference in CD8+ T-cell responses with remiquimod.48 Further studies using TLR agonists as adjuvants for tumor vaccines are also being conducted (Table 1).47-50
Inhibiting Immune Suppression
Tumors have mechanisms of suppressing immune responses in their microenvironment through checkpoint pathways, such as PD-1, and by means of suppressive cells, such as Tregs and MDSCs. Inhibiting the appropriate suppressive signals can improve immune surveil- lance and mount an antitumor T-cell response.
Immunosuppressive Cells
Tregs are among the strongest suppressors of the immune response within a tumor microenvironment, and their inhibition has been an active area of research. Researchers have been investigating the targeting of Treg-activating molecules on their surface through antibodies and vaccines. Checkpoint antibodies, LAG-3 and TIM-3, which are promising therapeutic targets, are discussed below. A phase I/II clinical trial of a dendritic cell vaccine with daclizumab, the anti-CD25 monoclonal antibody, showed patients who received daclizumab had significantly reduced Tregs; however, there was no improvement in PFS with daclizumab, perhaps because the Treg-depleted patients demonstrated fewer vaccine-specific effector T cells.51 Tyrosine kinase inhibitors already in clinical use, specifically sorafenib, sunitinib and imatinib, have demonstrated a decrease in Tregs, though their clinical efficacy against various malignancies varies.52-54 Thus, the proper use of Treg inhibition to obtain clinical benefit remains to be developed.
In addition to Tregs, infiltrative MDSCs act to further subdue the antitumor immune response. Certain chemotherapeutic agents, such as fluorouracil and gemcitabine, have been effective in killing MDSCs—although with low precision and significant toxicities—which may contribute to their responses in various solid tumors.55 Recently, results of a phase I trial demonstrated that an agonist antibody targeting the TRAIL R2 receptor effectively decreased MDSCs in 8 of 16 patients with advanced cancers, although with limited durability.56 Studies are underway exploring the value of combining checkpoint inhibitors and Treg and MDSC antibodies.
Inhibitory Checkpoints
Targeted inhibition of checkpoint molecules expressed on Tregs and CD8+ effector T cells in the tumor microenvironment—such as CTLA-4, PD-1, LAG-3, TIM-3, TIGIT, and BTLA—is one strategy that, in the case of CTLA-4 and PD-1, has thus far been successful. Further, LAG-3 has been found to play a particular role in antitumor responses in melanoma and ovarian cancer.57 In murine studies, inhibition of either TIM-3 or LAG-3, along with PD-1, led to prolonged immune responses and decreased tumor-specific Tregs in melanoma tumors, particularly in relapsed tumors.57,58 Novel PD-1 and CTLA-4 inhibitors are also being developed for a variety of cancers; there are 343 open trials on clinicaltrials.gov testing PD-1 inhibition in cancer. Various combinations of checkpoint-targeting agents are also being investigated in pre-clinical and clinical trials (Table 2).59-71
Another immunosuppressive molecule that has been under investigation for therapeutic intervention is indoleamine 2,3-dioxygenase 1 (IDO1), an enzyme produced in macrophages within the tumor microenvironment that acts on tryptophan metabolism. IDO1 decreases proliferation of T cells and increases neovascularization by countering interferon gamma.72 In a phase I clinical trial of 52 patients with metastatic solid tumors treated with epacadostat, an IDO1 inhibitor, 7 patients had stable disease, but no objective responses were seen.72 Currently, trials of combinations of IDO1 inhibitors with other immunomodulatory agents are ongoing, including a phase III trial in melanoma with the PD-1 inhibitor pembrolizumab.73
Integrins and Associated Proteins
Integrin proteins are yet another growing therapeutic area of interest in oncology. Integrins are transmembrane proteins expressed on most cells that facilitate communication between cells and their extracellular environment and control proliferation, survival, migration, and adhesion of cells. In cancer, the unique microenvironment surrounding the tumor, often hypoxic and deprived of nutrients compared with normal tissues, leads to increased expression of specific integrins that benefit the tumor.74 In addition to improving tumor survival and proliferation, certain integrins suppress immune cells in the tumor microenvironment. For example, TGF-β in the tumor microenvironment leads to increased expression of the integrin alpha E beta 7 (αEβ7) on Tregs, which promotes their suppressive function and localization.75 Interestingly, αEβ7 is also expressed on a variety of tumor-infiltrating lymphocytes, and pre-clinical studies have shown that when E-cadherin interacts with αEβ7, cytotoxic T cells are able to deliver toxic granules to cancer cells, which leads to apoptosis.
Loss of membrane E-cadherin has been associated with cancer spread and a poor prognosis in a variety of cancers, and re-instating E-cadherin in a tumor microenvironment may lead to increased immune targeting of the tumor.76 Other integrins, such as α4β 1 and integrin β3, play a role in recruitment and adhesion of leukocytes to tumors; integrin αvβ6 increases TGF-β, which suppresses T-cell activation and increases tumor proliferation; and integrins alpha v beta 3 (αvβ3) and αvβ5 (αvβ5) are associated with angiogenesis.
Currently, several monoclonal antibodies targeting integrins are in clinical trials.77 Etaracizumab, a monoclonal antibody against integrin αvβ3 has shown efficacy in a phase I trial in a variety of tumors, although a subsequent phase II trial showed no improvement in PFS or OS with etaracizumab.78 Cilengitide, an inhibitor of αvβ3 and αvβ5, showed efficacy and tolerability in phase I and phase II clinical trials in high-grade gliomas, with 69% having PFS at 6 months79,80; a phase III trial of cilengitide in patients with O[6]-methylguanine-DNA methyltransferase (MGMT) promoter methylated glioblastomas did not show an improvement in out- comes, however.81 Understanding the role of integrins and altering their expression to decrease immune suppression is an active area in immunotherapy research.
In addition to integrins, other molecules involved in cellular signaling that suppress immune function in the tumor microenvironment are being investigated as therapeutic targets. CD47, also known as integrin-associated protein, is a cell–surface membrane receptor protein found on many leukocytes. CD47 binds with beta-3 integrin, thrombospondin-1, signal regulatory protein-alpha (SIRP-α), and other signaling proteins to regulate T-cell activation, cell migration, phagocytosis, and other immune cell functions.
Specifically, CD47 bound to SIRP-α creates an inhibitory signal for phagocytosis often employed in tumor microenvironments. CD47 is expressed in many tumors and, interestingly, in cancer stem cells; it is thought expression of CD47 allows cancer stem cells to survive without being targeted by the immune system and that this leads to late-cancer recurrences.82 Targeting CD47 with monoclonal antibodies in murine models has shown to effectively treat acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), and leiomyosarcoma. Currently, multiple phase 1 trials are investigating CD47 inhibition in human patients with ALL, AML, and several solid tumors as described in Table 2.83-88
Adoptive T-cell Therapy
Another direction of immunotherapy is adoptive T-cell therapy, in which autologous T cells are harvested, engineered to recognize tumor antigens, and returned to the patient to specifically target the tumor. Chimeric antigen receptor (CAR) T cells have been developed to hold a membrane receptor that binds to a specific tumor antigen and contains an intracellular component that activates the T cell in the presence of the antigen.89 More recently, CAR-T cells have included a costimulatory signal, such as CD28 or CD137, that maintains activated T cells and may lead to a sustained immune response.90
CAR-T cells have also been engineered to express multiple CARs that recognize several tumor antigens and stimulate production of cytokines and interleukins when bound to their antigen. CAR-T cells are also being developed to alter pathways used by the tumor to suppress the immune system, such as PD-1. CARs targeting PD-1 have been introduced into T cells, with a mechanism that leads to T-cell activation signaling upon binding PD-1, with the goal of reversing immune suppression in the tumor.
Clinical trials are underway for many cancer types and antigens. The most success to date has been seen with hematologic malignancies, with several durable complete remissions seen in ALL, chronic lymphocytic leukemia, and lymphomas. In solid tumors, localization of T cells to the tumor has been challenging; however, trials targeting solid tumor antigens, such as CEA, ERBB2, VEGFR2, and a variety of others are being studied.90 A search on clinicaltrials.gov yielded over 160 open clinical trials with CAR-T cells.
Discussion and Conclusions
A greater understanding of the complex interactions between tumors and the immune system has led to a vast array of potential new therapeutic targets. Although many new agents show efficacy in vitro that demonstrate their desired function, turning these in vitro successes into clinical benefits has been challenging. Different tumors, even with the same underlying histology, show a wide range of heterogeneity in how they suppress the immune system, with tumors variably expressing PD-1 or having different quantities of infiltrating lymphocytes.91-93
Taking advantage of a tumor’s unique immune-altering mechanisms could allow for tailored immunotherapy for specific tumors. For example, metastatic melanoma tumors have been associated with strong inhibition of the STING pathway, leading to a de- creased immune response to cytosolic DNA, as would be present in a viral infection, and frequently express PD-1 and CTLA-4 on cells in its microenvironment. For these reasons, melanoma is very susceptible to T-VEC, as well as CTLA-4 and PD-1 inhibitors.94 Challenges in diagnostic testing have also complicated the tailored approach to immunotherapy in cancer. Markers of immune suppression that would be helpful for therapeutic implications, such as PD-1, are heterogeneously expressed throughout a tumor and change dynamically in response to stimuli.94,95 Thus, more research and trials need to take place to better understand the factors that lead to expression of dynamic markers and to develop accurate diagnostic tests for clinical use.
Discovering the appropriate combination of drugs to use against a specific tumor that affect the pathways and cells most active in that tumor represent the challenges of using these new technologies.
As our understanding of the tumor microenvironment continues to grow and we develop more accurate and clinically useful assays to describe the state of the tumor microenvironment, perhaps these new therapies will be tools used to individualized treatment regimens to reach meaningful clinical outcomes.
Financial disclosures: None.
Author affiliations: Juraj Kavecansky and Anna C. Pavlick are with New York University School of Medicine.
Address correspondence to: Juraj Kavecansky, MD, New York University School of Medicine, 550 First Ave., New York, NY 10016.
References
- Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17(11):1558-1568. doi: 10.1016/S1470-2045(16)30366-7.
- Borghaei H, Brahmer J, Horn L, et al. P2.35: nivolumab vs docetaxel in advanced NSCLC: CheckMate 017/057 2-Y update and exploratory cytokine profile analysis: track: immunotherapy. J Thorac Oncol. 2016;11(10S):S237-S238. doi: 10.1016/j. jtho.2016.08.106.
- Motzer RJ, Escudier B, McDermott DF, et al; CheckMate 025 investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803-1813. doi: 10.1056/ NEJMoa1510665.
- Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856-1867.
- Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311-319. doi: 10.1056/NEJMoa1411087.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi: 10.1016/j.cell.2011.02.013.
- Balkwill F, Charles KA, Mantovani A. Smoldering and polariz- ing inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211-217.
- Martin F, Apetoh L, Ghiringhelli F. Role of myeloid-derived suppressor cells in tumor immunotherapy. Immunotherapy. 2012;4(1):43-57. doi: 10.2217/imt.11.154.
- Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24(6):302-306.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253-268. doi: 10.1038/nri3175.
- Baksh K, Weber J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin Oncol. 2015;42(3):363-377. doi: 10.1053/j.seminoncol.2015.02.015.
- Wang C, Lin GH, McPherson AJ, Watts TH. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev. 2009;229(1):192-215. doi: 10.1111/j.1600- 065X.2009.00765.x.
- Alfaro C, Echeveste JI, Rodriguez-Ruiz ME, et al. Functional expression of CD137 (4-1BB) on T helper follicular cells. Oncoimunology. 2015;4(12):e1054597.
- Chester C, Ambulkar S, Kohrt HE. 4-1BB agonism: adding the accelerator to cancer immunotherapy. Cancer Immunol Immu- nother. 2016;65(10):1243-1248. doi: 10.1007/s00262-016-1829-2.
- Srivastava RM, Trivedi S, Concha-Benavente F, et al. CD137 stimulation enhances cetuximab-induced natural killer: dendritic cell priming of antitumor T-cell immunity in patients with head and neck cancer. Clin Cancer Res. 2017;23(3):707-716. doi: 10.1158/1078-0432.CCR-16-0879.
- Massarelli E, Segal N, Ribrag V, et al. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at: 31st Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
- Curti BD, Kovacsovics-Bankowski M, Morris N, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73(24):7189-7198. doi: 10.1158/0008- 5472.CAN-12-4174.
- Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: Structure and function - what questions remain? Mol Immunol. 2017;83:13- 22. doi: 10.1016/j.molimm.2017.01.006.
- Dubensky TW Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther Adv Vaccines. 2013;1(4):131-143. doi: 10.1177/2051013613501988.
- Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. 2016;37(12):855-865. doi: 10.1016/j. it.2016.09.006.
- Mohammed S, Bakshi N, Chaudri N, Akhter J, Akhtar M. Cancer vaccines: past, present, and future. Adv Anat Pathol. 2016;23(3):180-191. doi: 10.1097/PAP.0000000000000116.
- Kantoff PW, Higano CS, Shore ND, et al; IMPACT study investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411-422. doi: 10.1056/ NEJMoa1001294.
- Lesterhuis WJ, Schreibelt G, Scharenborg NM, et al. Wild- type and modified gp100 peptide-pulsed dendritic cell vaccination of advanced melanoma patients can lead to long-term clinical responses independent of the peptide used. Cancer Immunol Immu- nother. 2011;60(2):249-260. doi: 10.1007/s00262-010-0942-x.
- Lowenfeld L, Mick R, Datta J, et al. Dendritic cell vaccination enhances immune responses and induces regression of HER2pos DCIS independent of route: results of randomized selection design trial [ePub ahead of print]. Clin Cancer Res. 2016. pii: clincanres.1924.2016.
- Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel vaccines for glioblastoma: clinical update and perspective. Immunotherapy. 2016;8(11):1293-1308.
- Pfizer. Study of OX40 agonist PF-04518600 alone and in combination with 4-1BB agonist PF-0582566 (NCT02315066). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/ NCT02315066?term=NCT02315066&rank=1. Accessed February 8, 2017.
- Bauer TM, Chae YK, Patel S, et al. A phase I study of MEDI6383, an OX40 agonist, in adult patients with select advanced solid tumors. J Clin Oncol. 2015;33(suppl; abstr TPS3093).
- Powderly JD, Gutierrez M, Wang D, et al. A phase 1b/2, open-label study to evaluate the safety and tolerability of MEDI6469 in combination with immune therapeutic agents or therapeutic mAbs in patients with selected advanced solid tumors or aggressive B-cell lymphomas. J Clin Oncol. 2015;33(suppl; abstr TPS3091).
- Bell R, MedImmune LLC. Anti-OX40 antibody in head and neck cancer patients (NCT02274155). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02274155?ter- m=NCT02274155&rank=1. Accessed February 8, 2017.
- Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein pep- tide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. 2014;16(2):274-249. doi: 10.1093/ neuonc/not203.
- Wood L. Ad/HER2/Neu dendritic cell cancer vaccine test- ing (NCT01730118). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT01730118?term=NCT01730118&rank=1. Accessed February 8, 2017.
- Becerra C. Dendritic cell vaccine and chemotherapy for patients with pancreatic cancer (PancVax) (NCT02548169). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/ NCT02548169?term=NCT02548169&rank=1. Accessed February 8, 2017.
- Harrington KJ, Andtbacka RH, Collichio F, et al. Efficacy and safety of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in patients with stage IIIB/C and IVM1a melanoma: subanalysis of the phase III OPTiM trial. Onco Targets Ther. 2016;9:7081-7093.
- Loskog A. LOAd703 oncolytic virus therapy for pancreatic cancer (NCT02705196) Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02705196?term=NCT02705196&rank=1. Accessed February 8, 2017.
- Kelly K. Safety and effect of GL-ONC1 administered IV with or without eculizumab prior to surgery for patients with solid organ cancers undergoing surgery (NCT02714374) Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02714374?ter- m=NCT02714374&rank=1. Accessed February 8, 2017.
- Cassier P, Bouhamama A, Eberst L, Pilleul F, Terret C, Mastier C. Immunization strategy with intra-tumoral injections of Pexa-Vec with ipilimumab in metastatic/advanced solid tumors (NCT02977156). Clinicaltrials.gov website. https://clinicaltrials. gov/ct2/show/NCT02977156?term=NCT02977156&rank=1. Accessed February 8, 2017.
- PsiOxus Therapeutics Ltd. Phase I/II study of enadenotucirev intraperitoneally in ovarian cancer patients (NCT02028117). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/ NCT02028117?term=NCT02028117&rank=1. Accessed February 8, 2017.
- Burke J. SillaJen, Inc. Hepatocellular carcinoma study comparing vaccinia virus based immunotherapy plus sorafenib vs sorafenib alone (PHOCUS) (NCT02562755). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02562755?ter- m=NCT02562755&rank=1. Accessed February 8, 2017.
- Salazar LG, Lu H, Reichow JL, et al. Topical imiquimod plus nab-paclitaxel for breast cancer cutaneous metastases: a phase 2 clinical trial [ePub ahead of print]. JAMA Oncol. 2017. doi: 10.1001/jamaoncol.2016.6007.
- Sabado RL, Pavlick A, Gnjatic S, et al. Resiquimod as an immunologic adjuvant for NY-ESO-1 protein vaccination in patients with high-risk melanoma. Cancer Immunol Res. 2015;3(3):278-287. doi: 10.1158/2326-6066.CIR-14-0202.
- Leiberman F. Imiquimod and tumor lysate vaccine immunotherapy in adults with high risk or recurrent grade II gliomas (NCT01678352). Clinicaltrials.gov website. https://clinicaltrials. gov/ct2/show/NCT01678352?term=NCT01678352&rank=1. Accessed February 8, 2017.
- Adams S, Novik Y, Speyer J, et al. Toll-like receptor (TLR) 7 agonist, cyclophosphamide, and radiotherapy for breast cancer with skin metastases (NCT01421017). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT01421017?ter- m=NCT01421017&rank=1. Accessed February 9, 2017.
- Saha D, Wakimoto H, Rabkin SD. Oncolytic herpes simplex virus interactions with the host immune system. Curr Opin Virol. 2016;21:26-34. doi: 10.1016/j.coviro.2016.07.007.
- Puzanov I, Milhem MM, Minor D, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619-2626. doi: 10.1200/JCO.2016.67.1529.
- Markert JM, Razdan SN, Kuo HC, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22(5):1048-1055. doi: 10.1038/mt.2014.22.
- Desjardins A, Sampson JH, Peters KP, et al. Patient survival on the dose escalation phase of the oncolytic polio/rhinovirus recombinant (PVSRIPO) against WHO grade IV malignant glioma (MG) clinical trial compared to historical controls [abstract]. J Clin Oncol. 2016;36(suppl). Abstract 2061.
- Ohkuri T, Ghosh A, Kosaka A, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2(12):1199- 1208. doi: 10.1158/2326-6066.CIR-14-0099.
- Salmon H, Idoyaga J, Rahman A, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44(4):924-938. doi: 10.1016/j.immuni.2016.03.012.
- Lu H, Wagner WM, Gad E, et al. Treatment failure of a TLR- 7 agonist occurs due to self-regulation of acute inflammation and can be overcome by IL-10 blockade. J Immunol. 2010;184(9):5360- 5367. doi: 10.4049/jimmunol.0902997.
- Jacobs JF, Punt CJ, Lesterhuis WJ, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010;16(20):5067-5078. doi: 10.1158/1078-0432. CCR-10-1757.
- Desar IM, Jacobs JH, Hulsbergen-vandeKaa CA, et al. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 2011;129(2):507-512. doi: 10.1002/ijc.25674.
- Adotevi O, Pere H, Ravel P, et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J Immunother. 2010;33(9):991-998. doi: 10.1097/CJI.0b013e3181f4c208.
- Larmonier N, Janikashvili N, LaCasse CJ, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL-tumors. J. Immunol. 2008;181(10):6955–6963.
- Albeituni SH, Ding C, Yan J. Hampering immune suppressors: therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J. 2013;19(6):490-501. doi: 10.1097/ PPO.0000000000000006.
- Dominguez G, Condamine TC, Mony S, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res [ePub ahead of print]. 2016. pii: clincanres.1784.2016.
- Camisaschi C, Vallacchi V, Vergani E, et al. Targeting immune regulatory networks to counteract immune suppression in cancer. Vaccines (Basel). 2016;4(4). pii: E38.
- Goding, SR, Wilson, KA, Xie, Y, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. 2013;190(9):4899-4909. doi: 10.4049/jim- munol.1300271.
- Woo, SR, Turnis, ME, Goldberg, MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917-927. doi: 10.1158/0008-5472.CAN-11-1620.
- Overman M; MedImmune LLC. Tremelimumab (Anti-CT- LA-4) plus MEDI4736 (Anti-PD-L1) in resectable colorectal cancer liver metastases (NCT02754856). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02754856?ter- m=NCT02754856&rank=1. Accessed February 8, 2017.
- Agenus, Inc. AGEN-1884, an anti-CTLA-4 antibody, in advanced solid cancers (NCT02694822). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02694822?ter- m=NCT02694822&rank=1. Accessed February 8, 2017.
- Greten T. Tremelimumab with chemoembolization or ablation for liver cancer (NCT01853618). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT01853618?ter- m=NCT01853618&rank=1. Accessed February 8, 2017.
- Ott P, Rodig S. Ipilimumab with or without dabrafenib, trametinib, and/or nivolumab in treating patients with melanoma that is metastatic or cannot be removed by surgery (NCT01940809). Clinicaltrials.gov website. https://clinicaltrials. gov/ct2/show/NCT01940809?term=NCT01940809&rank=1. Accessed February 8, 2017.
- Santa-Maria C. MEDI4736 and tremelimumab in treat- ing patients with metastatic HER2 negative breast cancer (NCT02536794). Clinicaltrials.gov website. https://clinicaltrials. gov/ct2/show/NCT02536794?term=NCT02536794&rank=1. Accessed February 8, 2017.
- Makker V. A study of durvalumab with or without tremelimumab in endometrial cancer (NCT03015129). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT03015129?ter- m=NCT03015129&rank=1. Accessed February 8, 2017.
- Genentech, Inc. A study of atezolizumab (MPDL3280A) as first-line monotherapy for advanced or metastatic non-small cell lung cancer (NSCLC): clinical evaluation of novel blood-based diagnostics (B-F1RST) (NCT02848651) Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02848651?ter- m=NCT02848651&rank=1. Accessed February 8, 2017.
- Kurtz JE; ARCAGY/GINECO Group. ATALANTE: Atezolizumab vs placebo phase III study in late relapse ovarian cancer treated with chemotherapy + bevacizumab (ATALANTE) (NCT02891824). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02891824?term=NCT02891824&rank=1. Accessed February 8, 2017.
- PETHEMA Foundation. Pembrolizumab (MK-3475) in MM patients with residual disease (NCT02636010). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02636010?term=NCT02636010&rank=1. Accessed February 8, 2017.
- Beatty GL, O’Dwyer PJ, Clark J, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1epacadostat (INCB024360) in patients with advanced solid malignancies [ePub ahead of print]. Clin Cancer Res. 2017. pii: clincanres.2272.2016. doi: 10.1158/1078-0432.CCR-16-2272.
- Jones M; Incyte Corporation. A phase 3 study of pembrolizumab + epacadostat or placebo in subjects with unresectable or metastatic melanoma (Keynote-252/ECHO-301). (NCT02752074). Clinicaltrial.gov website. https://clinicaltrials. gov/ct2/show/NCT02752074?term=NCT02752074&rank=1. Accessed February 8, 2017.
- Grossman S, Lim M. Anti-LAG-3 or urelumab alone and in combination with nivolumab in treating patients with recurrent glioblastoma (NCT02658981). Clinicaltrial.gov website. https://clinicaltrials.gov/ct2/show/NCT02658981?term=NCT02658981&rank=1. Accessed February 7, 2017.
- Bristol-Myers Squibb. Safety study of anti-LAG-3 in re- lapsed or refractory hematologic malignancies (NCT02061761). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02061761?term=NCT02061761&rank=1. Accessed February 7, 2017.
- Tesaro. A phase I study of TSR-022, an anti-TIM-3 mono- clonal antibody, in patients with advanced solid tumors (NCT02817633). Clinicaltrials.gov website. https://clinicaltrials. gov/ct2/show/NCT02817633?term=NCT02817633&rank=1. Accessed February 7, 2017.
- Novartis. Safety and efficacy of MBG453 as single agent and in combination with PDR001 in patients with advanced malignancies (NCT02608268). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02678338?ter- m=NCT02678338&rank=1. Accessed February 7, 2017.
- Vyas P, Chao M. CAMELLIA: Anti-CD47 antibody therapy in relapsed/refractory acute myeloid leukaemia (NCT02678338). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/ NCT02678338?term=NCT02678338&rank=1. Accessed February 8, 2017.
- Burgess M. Celgene. A phase I, dose finding study of CC- 90002 in subjects with advanced solid and hematologic cancers (NCT02367196). Clinicaltrials.gov website. https://clinicaltrials. gov/ct2/show/NCT02367196?term=NCT02367196&rank=1. Accessed February 8, 2017.
- Sievers E, Trillium Therapeutics. A trial of TTI-621 for patients with hematologic malignancies (NCT02663518). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02663518?ter- m=NCT02663518&rank=1. Accessed February 8, 2017.
- Takimoto C; Forty Seven, Inc. Phase I trial of HU5F9-G4, a CD47-targeting antibody (NCT02216409). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02216409?ter- m=NCT02216409&rank=1. Accessed February 8, 2017.
- Takimoto C; Forty Seven, Inc. Trial of Hu5F9-G4 in combination with cetuximab in patients with solid tumors and advanced colorectal cancer (NCT02953782). Clinicaltrials.gov website. https://clinicaltrials.gov/ct2/show/NCT02953782?ter- m=NCT02953782&rank=1. Accessed February 8, 2017.
- Ata R, Antonescu CN. Integrins and cell metabolism: an intimate relationship impacting cancer. Int J Mol Sci. 2017;18(1): pii: E189. doi: 10.3390/ijms18010189.
- Hadley GA, Higgins JM. Integrin αEβ7: molecular features and functional significance in the immune system. Adv Exp Med Biol. 2014;819:97-110. doi: 10.1007/978-94-017-9153-3_7.
- Le Floc’h A, Jalil A, Vergnon I, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. 2007;204(3):559-570.
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9-22. doi: 10.1038/nrc2748.
- Delbaldo C, Raymond E, Vera K, et al. Phase I and pharmaco- kinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against alphavbeta3 integrin receptor, in patients with advanced solid tumors. Invest New Drugs. 2008;26(1):35-43.
- Hersey P, Sosman J, O’Day S, et al; Etaracizumab Melanoma Study Group. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or - dacarbazine in patients with stage IV metastatic melanoma. Cancer. 2010;116(6):1526-1534. doi: 10.1002/cncr.24821.
- Stupp R, Hegi ME, Neyns B, et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(16):2712-2718. doi: 10.1200/JCO.2009.26.6650.
- Stupp R, Hegi ME, Gorlia T, et al; European Organisation for Research and Treatment of Cancer (EORTC); Canadian Brain Tumor Consortium; CENTRIC study team. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EO- RTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1100-1108. doi: 10.1016/S1470-2045(14)70379-1.
- Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271-285. doi: 10.1016/j. cell.2009.05.046.
- Liu X, Kwon H, Li Z, Fu YX. Is CD47 an innate immune checkpoint for tumor evasion? J Hematol Oncol. 2017;10(1):12. doi: 10.1186/s13045-016-0381-z.
- de Witte MA, Kierkels GJ, Straetemans T, Britten CM, Kuball J. Orchestrating an immune response against cancer with engineered immune cells expressing αβTCRs, CARs, and innate immune receptors: an immunological and regulatory challenge. Cancer Immunol Immunother. 2015;64(7):893-902. doi: 10.1007/s00262-015-1710-8.
- Holzinger A, Barden M, Abken H. The growing world of CAR T cell trials: a systematic review. Cancer Immunol Immuno- ther. 2016;65(12):1433-1450.
- Sasada T, Suekane S. Variation of tumor-infiltrating lymphocytes in human cancers: controversy on clinical significance. Immunotherapy. 2011;3(10):1235-1251. doi: 10.2217/ imt.11.106.
- McLaughlin J, Han G, Schalper KA, et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non– small-cell lung cancer. JAMA Oncol. 2016;2(1):46-54. doi: 10.1001/jamaoncol.2015.3638.
- Scognamiglio G, De Chiara A, Di Bonito M, et al. Variability in immunohistochemical detection of programmed death ligand 1 (PD-L1) in cancer tissue types. Int J Mol Sci. 2016;17(5). pii: E790. doi: 10.3390/ijms17050790.
- Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76(22):6747-6759. doi: 10.1158/0008- 5472.CAN-16-1404.
- Rehman JA, Han G, Carvajal-Hausdorf DE, et al. Quantitative and pathologist-read comparison of the heterogeneity of programmed death-ligand 1 (PD-L1) expression in non- small cell lung cancer. Mod Pathol. 2016. doi: 10.1038/mod- pathol.2016.186.